























El neuroblastoma es el tumor extracraneal sólido más frecuente en la infancia1, y el cuarto en frecuencia en el cómputo total de neoplasias infantiles después de las leucemias, los tumores del sistema nervioso central (SNC) y linfomas. Representa el 7%2 de todos los cánceres pediátricos y es la causa del 15% del total de muertes por procesos oncológicos en la infancia.
Presenta un amplio espectro de comportamiento, ya que se trata de la neoplasia en que se han demostrado más casos de regresión espontánea y diferenciación a tumor benigno, mientras que en niños con formas metastásicas puede causar el fallecimiento.
En los últimos años se ha desarrollado una importante investigación para predecir el riesgo de recaída al diagnóstico, lo que ha permitido clasificar a los pacientes en grupos pronósticos para adaptar la intensidad del tratamiento al “riesgo”. Al aplicar estos principios, la supervivencia de los niños con neuroblastoma ha aumentado en las últimas décadas3.
Puntos clave

La incidencia del neuroblastoma oscila entre 8 y 10 casos por millón de niños y año1. Se han descrito casos familiares de neuroblastoma, pero son raros; en estos casos, se transmite de forma autosómica dominante con penetrancia incompleta. Se ha observado una frecuencia mayor en pacientes afectados de neurofibromatosis y síndrome de Beckwith-Wiedeman. Se desconoce la etiología del neuroblastoma.
Presentación clínicaEl neuroblastoma puede originarse a lo largo de toda la cadena simpática, por lo que la localización del tumor primitivo puede ser muy variable. Las localizaciones más frecuentes son el abdomen (69%), especialmente en niños mayores, y el mediastino (21%), más importante en lactantes. Un 1% de los casos se diagnostican por las metástasis sin que se pueda localizar el tumor primitivo.
La mayoría de los diagnósticos se realizan en menores de 5 años, y su detección es rara después de 10 años.
Las metástasis se producen por vía hematológica y linfática y están presentes en el 43% de los niños al diagnóstico, especialmente en los mayores de 1 año, siendo las más frecuentes en la medula ósea, el hueso, el hígado y la piel. La sintomatología depende de la compresión e infiltración de órganos vecinos, de las metástasis y, en ocasiones, de síndromes paraneoplásicos.
En el lactante, es típico el descubrimiento casual de una masa abdominal o torácica asintomáticas en un examen sistemático de salud. En el niño mayor, es más frecuente el síndrome de “malestar maligno”, caracterizado por palidez, anemia, dolor de extremidades y febrícula.
Los tumores paraespinales pueden producir cuadros de compresión medular por invasión del canal a través de los agujeros de conjunción. El cuadro es más frecuente en neuroblastomas torácicos y en lactantes, y puede llegar a causar una paraplejia completa si no se diagnostican y tratan a tiempo.
Los tumores cervicales o mediastínicos pueden acompañarse de síndrome de Horner, consistente en ptosis palpebral, miosis y anhidrosis del lado afectado por lesión del ganglio estrellado.
En algunos casos, pueden presentar taquicardia, rubor facial e hipertensión arterial mediada por secreción de catecolaminas o mediada por renina, por afectación de los vasos renales por infiltración tumoral.
El neuroblastoma metastásico puede producir sintomatología según los órganos diana:
- —
La afectación masiva del hígado es particularmente frecuente en lactantes (estadio 4S) (tabla 1) y produce gran distensión abdominal por la enorme hepatomegalia. En casos extremos, dificulta el retorno venoso, y se produce edema del escroto y las extremidades inferiores, así como dificultad respiratoria.
Tabla 1.Sistema Internacional de Estadificación del Neuroblastoma (INSS)
El INSS combina ciertas características de los sistemas Pediatric Oncology Group y Children's Cáncer Group que se empleaban anteriormente y ha identificado a grupos con pronósticos definidos Estadio 1. Tumor localizado con escisión macroscópica completa con enfermedad residual microscópica o sin ésta; ganglios linfáticos ipsilaterales representativos microscópicamente negativos para el tumor (como los nódulos adheridos al tumor primario y extirpados junto con éste pueden ser positivos)
Estadio 2A. Tumor localizado con escisión macroscópica incompleta; ganglios linfáticos ipsilaterales representativos negativos para el tumor microscópicamente
Estadio 2B. Tumor localizado con escisión macroscópica completa o sin ésta; ganglios linfáticos ipsilaterales positivos para el tumor. Los ganglios linfáticos contralaterales agrandados deben ser negativos microscópicamente
Estadio 3. Tumor irresecable unilateral infiltrante más allá de la línea media con afectación de los ganglios linfáticos regionales o sin ésta; o tumor unilateral localizado con afectación de los ganglios linfáticos regionales contralaterales; o tumor en la línea media con extensión bilateral por infiltración (irresecable) o por afectación del ganglio linfático. La línea media está determinada por la columna vertebral
Estadio 4. Todo tumor primario con diseminación a ganglios linfáticos distantes huesos la médula ósea hígado piel u otros órganos con excepción de lo definido para el estadio 4S
Estadio 4S. Tumor primario localizado como se define para el estadio 1 2A o 2B con diseminación limitada a la piel el hígado o la médula ósea. La afectación medular debe ser mínima (o sea < 10% de células nucleadas totales identificadas como malignas por biopsia de hueso o por aspirado de médula ósea). Una afectación más extensa de la médula ósea se consideraría como enfermedad en estadio IV. Los resultados de la gammagrafía con metayodobencilguanidina en caso de que se efectúe deben ser negativos para la enfermedad en la médula ósea
- —
Las metástasis subcutáneas (típicas del estadio 4S) (tabla 1) se manifiestan como nódulos múltiples duros, no dolorosos y, a veces, de coloración azulada.
- —
En el niño mayor con metástasis óseas, es frecuente la afectación de huesos periorbitarios y retrobulbar, causando proptosis y equimosis periorbitaria o “hematoma en anteojos”.
La afectación medular puede dar dolor óseo en la deambulación o irritabilidad en los lactantes. Hay algunos síndromes paraneoplásicos característicos de este tumor:
- 1.
El síndrome opsocerebelomioclónico, que parece tener un origen autoinmunitario, puede asociarse al neuroblastoma hasta en un 2-4% de los casos, y se caracteriza por irritabilidad continua, junto con movimientos incontrolados de los ojos en todas direcciones (opsoclonus) y, en ocasiones, también ataxia cerebelosa y/o mioclonías. Se suele dar en neuroblastomas localizados, sin amplificación del gen N-myc (NMA) y con buen pronóstico vital. Sin embargo, puede llevar a secuelas motoras y cognitivas, déficit en el lenguaje y trastornos del comportamiento a largo plazo4. Hay controversia actual sobre cuál debe ser el tratamiento, glucocorticoides frente a inmunoglobulinas intravenosas. También puede existir sin asociarse al neuroblastoma, pero casi en el 50% de ellos se descubrirá un neuroblastoma asociado.
- 2.
Un 7-9% son tumores secretores de péptido intestinal vasoactivo, por lo que presentan un cuadro de diarrea acuosa intratable, con hipopotasemia, hipocalcemia y deshidratación. La resección quirúrgica suele resolver completamente los síntomas.
Lectura rápida

El neuroblastoma es el tumor extracraneal sólido más frecuente en la infancia, presenta un amplio espectro de comportamiento, ya que puede madurar e involucionar en los casos perinatales, pero hay otros casos metastásicos con mala evolución, a pesar de tratamientos muy agresivos.
En los últimos años se ha desarrollado una investigación importante que ha hecho posible la estratificación de los pacientes en grupos de riesgo de recaída, que permiten adaptar la intensidad del tratamiento a ese riesgo.

Para confirmar el diagnóstico de neuroblastoma hay unos criterios consensuados5:
- —
Diagnóstico anatomopatológico del tejido tumoral con o sin inmunohistoquímica, microscopia electrónica o aumento de catecolaminas urinarias (o séricas) o sus metabolitos (ácido homovanílico, vanilmandélico o dopamina).
- —
Infiltración de la médula ósea (aspirado o biopsia) por células tumorales y aumento de la excreción urinaria de catecolaminas.
Para clasificar la extensión del neuroblastoma al diagnóstico, se creó el International Neuroblastoma Staging System (INSS), revisado en 1993 (tabla 1). El International Neuroblastoma Response Criteria (INRC) (tabla 2) se utiliza para valorar la respuesta6. Ambos surgieron por consenso entre los principales grupos de trabajo en neuroblastoma de Estados Unidos, Europa y Japón6,7.
Criterios internacionales de respuesta en neuroblastoma (INRC)
| Respuesta | Tumor primario | Metástasis |
|---|---|---|
| Completa | Sin tumor | Sin tumor; catecolaminas normales |
| Parcial muy buena | Disminución 90-99% | Sin tumor Se permiten cambios residuales en huesos en tecnecio 99 |
| Parcial | Disminución > 50% | Disminución > 50% en todas la localizaciones medibles Huesos y médula ósea: disminución > 50% del número de zonas positivas. Sólo se permite una muestra de médula ósea positiva |
| Mixta | Sin nuevas lesiones; reducción > 50% de cualquier lesión medible, con reducción < 50% de otras o aumento < 25% de alguna lesión preexistente | |
| Sin respuesta | Sin lesiones nuevas; reducción < 50% pero asociado a aumento < 25% de cualquier lesión preexistente | |
| Progresión | Aparición de cualquier lesión nueva; aumento > 25% de cualquier lesión medible; médula ósea que se hace positiva siendo negativa previamente | |
Así pues, la confirmación histológica se requiere para el diagnóstico definitivo. El tejido se obtiene del tumor primario o del aspirado/ biopsia de médula ósea en los niños que tienen afectación metastásica en médula ósea. Se deben realizar 2 aspirados y 2 biopsias de médula ósea para excluir la infiltración. En los lactantes, las biopsias se pueden sustituir por otros 2 aspirados.
- —
Ecografía: es muy sensible para los tumores abdominales, e incluye la detección de metástasis linfáticas y hepáticas; sin embargo, la tomografía computarizada (TC) y la resonancia magnética (RM) valoran mejor el volumen del tumor; además, esta última es superior para evaluar la invasión del canal medular. Es necesario realizar una radiografía de tórax, la RM/TC se realizarán en caso de masa o adenopatías que se extienden al tórax. En caso de clínica compatible con afectación cerebral, se realizará TC craneal.
- —
Gammagrafía ósea con metayodobencilguanidina (MIBG): es un isótopo radiactivo que sigue las mismas rutas metabólicas que las catecolaminas, y es captado específicamente por el tumor y sus metástasis en el 86% de los neuroblastomas, por lo que se recomienda en el diagnóstico y en las evaluaciones posteriores8. En los casos en que no hay captación en el tumor primitivo, debe realizarse gammagrafía con tecnecio 99 para excluir la presencia de metástasis óseas.
La determinación de catecolaminas en orina9 es uno de los métodos más fiables para el diagnóstico y el seguimiento del tumor durante y después del tratamiento. La sensibilidad y la especificidad de la técnica se aproxima al 90-95%. Puede verse influido por la dieta (vainilla, plátano) u otros fármacos.
Lectura rápida
El neuroblastoma puede originarse a lo largo de toda la cadena simpática, pero las localizaciones más frecuentes son el abdomen y el mediastino. Las metástasis más frecuentes son: médula ósea, hueso, hígado y piel
Es característico el descubrimiento casual de una masa abdominal o torácica asintomáticas en un examen habitual de salud en un lactante. En el niño mayor, es más frecuente el síndrome de “malestar maligno”, caracterizado por palidez, anemia, dolor de extremidades y febrícula.
La sintomatología depende de la compresión e infiltración de órganos vecinos, de las metástasis y, en ocasiones, de síndromes paraneoplásicos. Incluso en los casos localizados, puede ser necesario un tratamiento temprano, debido a sintomatología compresiva local (como parálisis en caso de afectación raquimedular o insuficiencia respiratoria en caso de afectación hepática).
El neuroblastoma forma parte del grupo de tumores de células azules, redondas, pequeñas típicas del niño. Se cree que procede de células primitivas del sistema simpático pluripotenciales. Hay 3 patrones histopatológicos clásicos que reflejan un espectro de maduración y diferenciación (tabla 3):
- —
Neuroblastoma típico: compuesto por células pequeñas, de tamaño uniforme, con núcleo denso hipercromático y escaso citoplasma. La presencia de prolongaciones neuríticas es patognomónica, las seudorrosetas de Homer-Wright son otro hallazgo frecuente.
- —
En el otro extremo, el ganglioneuroma es un tumor completamente diferenciado, compuesto de células ganglionares maduras, estroma y células de Schwan.
- —
El ganglioneuroblastoma es un tumor heterogéneo que presenta características de ambos tumores en diferente proporción. Se subclasifica en difuso o nodular, teniendo un peor pronóstico este último.
Shimada et al10 desarrollaron una clasificación histológica que, combinando la edad del paciente con otros factores, como el grado de diferenciación y el índice de mitosis-cariorrexis, ha demostrado tener valor pronóstico. Recientemente, un grupo de patólogos de Estados Unidos y Europa encabezados por Shimada la han refinado y mejorado, y la han convertido en la INPC (International Neuroblastoma Pathology Classification11), que es la clasificación histológica utilizada actualmente por todos los grandes grupos en sus estudios cooperativos.
Biología y enfermedad molecularLa NMA12 se da en el 20% de los neuroblastomas, sobre todo en formas avanzadas (33%) y en menor proporción en estadios 1, 2, 3 y 4S (9%). Se asocia a mal pronóstico por rápida progresión tumoral.
La deleción del 1p13 se encuentra en un 47% de neuroblastomas, especialmente en estadios avanzados, se asocia casi siempre con NMA y, en los tumores en que se presenta de forma aislada, se asocia con más riesgo de recidivas locales.
La ganancia de 17q es un cambio cromosómico frecuente en tumores neuroblásticos y que tiene significado pronóstico desfavorable, con independencia de otros factores14, aunque hay resultados controvertidos según el tamaño del fragmento cromosómico ganado.
Las alteraciones en el brazo q del cromosoma 11 también tienen un efecto pronóstico desfavorable en pacientes con tumores no metastásicos2.
Los tumores con índice de ADN aneuploide (especialmente triploide) suelen ser localizados y de mejor pronóstico que los diploides o tetraploides, especialmente en niños pequeños.
Cribado de neuroblastomaMás del 90% de los neuroblastomas excretan metabolitos de las catecolaminas en la orina; por otra parte, la edad es un factor pronóstico muy importante. Por ello, se han llevado a cabo estudios extensos15,16 de detección temprana mediante determinación de catecolaminas urinarias, pensando en que la rápida detección de la enfermedad en fase localizada podría disminuir la incidencia de enfermedad de alto riesgo o avanzada y así mejorar la supervivencia.
Pero los pacientes diagnosticados por cribado estaban habitualmente en estadios iniciales, sin NMA y tenían muy buen pronóstico, y los niños negativos en el cribado, en los que se diagnosticaba un neuroblastoma más tarde, presentaban estadios avanzados y con biología desfavorable. Es decir, los programas de cribado aumentaban la incidencia de neuroblastomas “favorables”, mientras que fallaban en reducir la incidencia de enfermedad avanzada desfavorable, por lo que no disminuían la mortalidad.
Estos datos indican que hay al menos 2 tipos distintos de neuroblastoma: uno con factores biológicos favorables y gran capacidad de regresión espontánea, que desaparecería en su mayoría sin necesidad terapéutica, y otro que afecta a niños mayores, con factores biológicos desfavorables.
Lectura rápida
El diagnóstico se confirma obteniendo tejido tumoral o mediante estudio de la médula ósea si hay infiltración.
La ecografía es un método muy sensible para los tumores abdominales, además es inocua y fácil de realizar. La resonancia magnética valora mejor la invasión del canal medular y la gammagrafía ósea con metayodobencilguanidina es muy sensible y específica para detectar metástasis, se utiliza para la estadificación y también para valorar la respuesta tumoral.
Para la clasificación de la extensión del neuroblastoma al diagnóstico, se utiliza el International Neuroblastoma Staging System (INSS), y para valorar la respuesta se usa el International Neuroblastoma Response Criteria (INRC).
Para el seguimiento, se puede utilizar la determinación de catecolaminas en orina que tiene una sensibilidad y una especificidad que se aproxima al 90-95%, también la metayodobencilguanidina es muy importante.
Factores pronósticosComo factores pronósticos se utilizan: edad, clasificación histopatológica o INPC (International Neuroblastoma Pathology Classification), factores de biología molecular, como la amplificación del gen N-myc, la deleción del 1p y la ploidía
Los factores pronósticos clásicos en el neuroblastoma eran la edad y el estadio, a los que en los últimos años se han añadido otros factores histológicos y biológicos del tumor que han permitido identificar subgrupos con diferente pronóstico vital17.
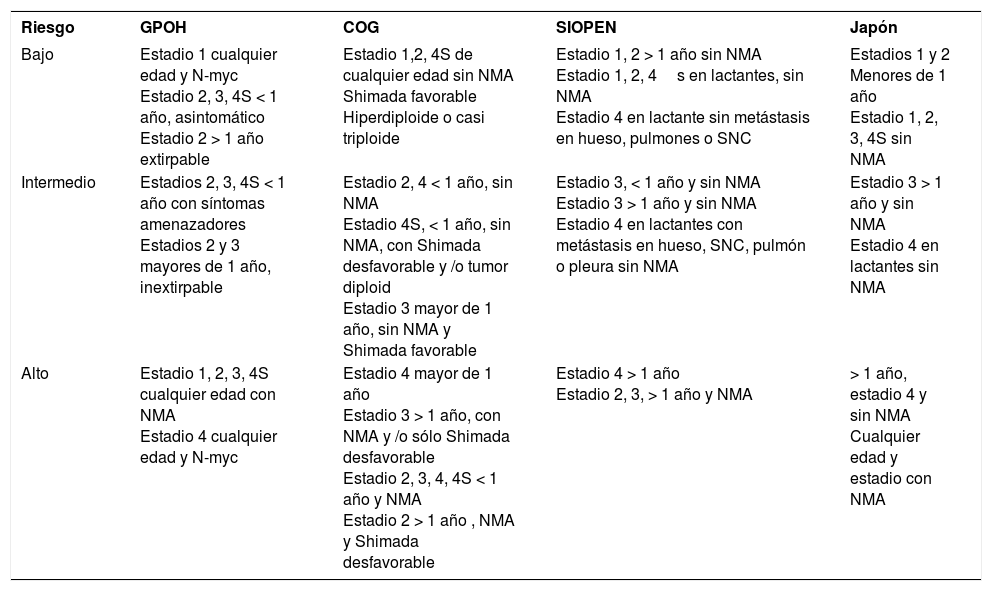
La importancia de este enfoque radica en la adaptación del tratamiento al “riesgo”18 que se realiza desde el diagnóstico y que ha permitido desescalar el tratamiento en las formas favorables e intensificarlo en los de mal pronóstico (tabla 4). Esto sólo se puede realizar con garantía si se ha estudiado completamente al paciente, incluidos estudios biológicos del tumor, para lo que es imprescindible realizar biopsia, y que ésta se estudie en un laboratorio de referencia o con amplia experiencia.
Comparación de la estadificación por riesgo en los diferentes grupos cooperativos
| Riesgo | GPOH | COG | SIOPEN | Japón |
|---|---|---|---|---|
| Bajo | Estadio 1 cualquier edad y N-myc Estadio 2, 3, 4S < 1 año, asintomático Estadio 2 > 1 año extirpable | Estadio 1,2, 4S de cualquier edad sin NMA Shimada favorable Hiperdiploide o casi triploide | Estadio 1, 2 > 1 año sin NMA Estadio 1, 2, 4s en lactantes, sin NMA Estadio 4 en lactante sin metástasis en hueso, pulmones o SNC | Estadios 1 y 2 Menores de 1 año Estadio 1, 2, 3, 4S sin NMA |
| Intermedio | Estadios 2, 3, 4S < 1 año con síntomas amenazadores Estadios 2 y 3 mayores de 1 año, inextirpable | Estadio 2, 4 < 1 año, sin NMA Estadio 4S, < 1 año, sin NMA, con Shimada desfavorable y /o tumor diploid Estadio 3 mayor de 1 año, sin NMA y Shimada favorable | Estadio 3, < 1 año y sin NMA Estadio 3 > 1 año y sin NMA Estadio 4 en lactantes con metástasis en hueso, SNC, pulmón o pleura sin NMA | Estadio 3 > 1 año y sin NMA Estadio 4 en lactantes sin NMA |
| Alto | Estadio 1, 2, 3, 4S cualquier edad con NMA Estadio 4 cualquier edad y N-myc | Estadio 4 mayor de 1 año Estadio 3 > 1 año, con NMA y /o sólo Shimada desfavorable Estadio 2, 3, 4, 4S < 1 año y NMA Estadio 2 > 1 año , NMA y Shimada desfavorable | Estadio 4 > 1 año Estadio 2, 3, > 1 año y NMA | > 1 año, estadio 4 y sin NMA Cualquier edad y estadio con NMA |
COG: Children's Oncology Group; GPOH: German Pediatric Oncology-Hematology; NMA: amplificación del gen N-myc; SIOPEN: European Neuroblastoma-International Society Pediatric Oncology; SNC: sistema nervioso central.
Aunque hay multitud de factores pronósticos conocidos, muchos de ellos no tienen un valor independiente y se presentan asociados con otros de más peso específico.
La edad, el estadio y la NMA son los factores admitidos por todos los grupos cooperativos. A ellos se añaden con menor valor la clasificación histológica de Shimada, la deleción del 1 p y la ploidía. Otros marcadores biológicos, como el receptor de la tirosincinasa (TRKA/B), la resistencia a quimioterapia (MDR1) y la ganancia del 17p, han demostrado valor pronóstico en estudios.
El límite de edad para separar formas de riesgo ha sido de 12 meses clásicamente; sin embargo, estudios recientes con series grandes con factores biológicos del tumor estudiados demuestran que su influjo favorable se ejerce hasta los 18–20 meses19.
Lectura rápida
El tratamiento se adapta según el grupo de riesgo.
La cirugía inicial se utiliza para establecer el diagnóstico, realizar estudios biológicos y ayudar a la estadificación del tumor. cuando hay datos de riesgo quirúrgico, se desaconseja la intervención con intento radical, y realizar cirugía retardada que evalúa la respuesta a la quimioterapia y extirpa todo el tumor residual posible con menor número de complicaciones.
Las modalidades de tratamiento usadas en el neuroblastoma son cirugía, quimioterapia y radioterapia, más modificadores de la respuesta biológica y/o inmunoterapia añadidos recientemente. La utilización de cada uno de ellos y su mayor o menor intensidad van a depender del grupo de riesgo al que se asigne el paciente, es decir, de la anticipación del comportamiento clínico que se espera que tenga cada caso individual.
CirugíaLa cirugía desempeña un papel básico tanto en el diagnóstico como en el tratamiento. Los objetivos de la cirugía inicial son establecer el diagnóstico, suministrar tejido para estudios biológicos20, ayudar a la estadificación del tumor e intentar extirparlo sin lesionar estructuras vitales. En el caso de la cirugía retardada, se evalúa la respuesta a la quimioterapia y se extirpa todo el tumor residual posible.
Las posibilidades de extirpación del tumor dependen de su localización, relación con los grandes vasos y características del propio tejido tumoral (friabilidad, hemorragia, etc.). El sacrificio de estructuras vitales debe evitarse en la cirugía diagnóstica, ya que la quimioterapia permite disminuir el tamaño del tumor primario y los ganglios linfáticos, y también cambia la consistencia, con lo que se favorecen extirpaciones retardadas con un número menor de complicaciones.
El grupo de neuroblastoma de la SIOP-Europa (SIOPEN) ha desarrollado unos criterios de riesgo quirúrgico basados en el estudio radiológico21. La presencia de cualquiera de los datos de riesgo desaconseja la intervención quirúrgica con intento radical por la gran frecuencia de complicaciones.
Se han publicado complicaciones quirúrgicas en el neuroblastoma entre el 5 y el 25%22. Las complicaciones son mucho menores en la cirugía retardada tras la quimioterapia. Las más frecuentes son nefrectomía, hemorragias operatorias, lesión de los vasos renales que conduce a atrofia renal y síndrome de Horner.
RadioterapiaEl neuroblastoma es un tumor radiosensible, su papel actual permanece en el tratamiento local de los pacientes de riesgo alto (estadio 4 en niños mayores de 1 año), especialmente con NMA, por la gran frecuencia de recaídas locales en estos pacientes.
QuimioterapiaEs el recurso terapéutico predominante en los pacientes con neuroblastoma de riesgo intermedio o alto. Estudios de fase II han demostrado la efectividad de los agentes alquilantes, derivados de platino, epipodofilotoxinas y doxorubicina. Se administran en esquemas combinados, se intenta maximizar su eficacia y disminuir los efectos secundarios a largo plazo.
Lectura rápida
La quimioterapia es un recurso terapéutico de gran valor en el neuroblastoma de riesgo intermedio o alto. Estudios de fase II han demostrado la efectividad de los agentes alquilantes, derivados de platino, epipodofilotoxinas y doxorubicina. Se administran en esquemas combinados intentando maximizar su eficacia y disminuir los efectos secundarios a largo plazo.
Masas adrenales congénitas: en la mayor parte de los casos se trata de pacientes con excelente pronóstico que pueden ser observados en espera de que regresen de forma espontánea.
Neuroblastoma de alto riesgo: con el uso de la quimioterapia de inducción, la cirugía y la megaterapia con trasplante autólogo, las tasas de respuesta oscilan entre el 60 y el 80% de todos los pacientes, pero muchos de estos pacientes recaen. Para intentar controlar este fenómeno, se están utilizando agentes diferenciadores, como los retinoides y los anticuerpos monoclonales. A pesar de un tratamiento muy agresivo, el pronóstico es muy malo en este grupo de pacientes, por lo que se siguen buscando otras alternativas.

Los pacientes se clasifican en grupos de riesgo aceptados por el ESIOP-neuroblastoma con recomendaciones terapéuticas diferentes para cada grupo (tabla 4).
Diagnóstico prenatalEl uso habitual de la ecografía durante el embarazo y en el período posnatal ha conducido al diagnóstico prenatal y posnatal de un número creciente de neuroblastomas localizados, cuyo manejo plantea dudas.
En la mayor parte de los casos, se trata de pacientes con pronóstico excelente que pueden ser observados en espera de la regresión espontánea.
Se consideran datos desfavorables: tamaño de la masa mayor de 5cm, aspecto necrótico e infiltración de órganos vecinos. En estos casos, se debe realizar extirpación quirúrgica temprana y, si ésta no es posible, biopsia para estudio de factores biológicos en el tumor y decidir tratamiento u observación con una base más sólida.
Grupo de buen pronósticoLos niños con neuroblastoma de estadio 1 o 2 de cualquier edad y el estadio 4S, todos ellos sin NMA, tienen un pronóstico excelente. Estos pacientes requieren un tratamiento mínimo, generalmente sólo cirugía, a excepción de los casos que presentan síntomas de compresión medular o bien afectación hepática que afecte a la vida.
El neuroblastoma en reloj de arena es más frecuente en niños con enfermedad localizada, especialmente en lactantes. El tratamiento de la compresión medular sintomática mediante administración de quimioterapia y dexametasona puede evitar la cirugía descompresiva y la radioterapia en algunos casos, siendo la laminotomía necesaria sólo en pacientes con deterioro neurológico muy rápido23.
La capacidad de regresión espontánea es una de las características típicas del estadio 4S del neuroblastoma; sin embargo, algunos casos presentan un comportamiento inicial muy agresivo que puede conducir al fallecimiento del paciente24. Este tipo de presentación es más frecuente en recién nacidos y lactantes menores de 2 meses al diagnóstico.
Se recomienda administrar quimioterapia a los pacientes con riesgo de muerte por insuficiencia hepática o respiratoria, mientras que el resto son vigilados de forma atenta. Los fármacos recomendados son el carboplatino/etopósido, administrados en un número mínimo de ciclos hasta conseguir detener la progresión rápida de la enfermedad y estabilizar al paciente.
Es necesario obtener tejido tumoral para estudiar factores biológicos, como la NMA, ya que, aunque es infrecuente en el estadio 4S, requiere un tratamiento intensivo, como los neuroblastomas de riesgo alto.
Los lactantes con neuroblastoma estadio 3 sin NMA tienen muy buen pronóstico con quimioterapia suave y cirugía diferida.
Grupo de pronóstico intermedioEstá compuesto por niños con neuroblastoma de estadio 3 sin NMA mayores de 1 año y estadio 4 sin amplificación en lactantes.
Este grupo alcanza resultados terapéuticos excelentes con cirugía y quimioterapia de intensidad moderada.
Las discusiones terapéuticas sobre el estadio 3 se centran en la posibilidad de extirpar el tumor primitivo al diagnóstico o tras quimioterapia. La tendencia actual es realizar una biopsia al diagnóstico, y resecar el tumor después de la quimioterapia de inducción, seguido de una quimioterapia de mantenimiento. De este modo, el 70-90% de los neuroblastomas de estadio 3 se convierten en extirpables y, además, se reduce el riesgo de complicaciones quirúrgicas.
Los fármacos usados de forma más habitual son: ciclofosfamida/vincristina, carboplatino y etopósido, y de forma menos frecuente doxorubicina y cisplatino.
El pronóstico de los lactantes con neuroblastoma de estadio 4 es mucho mejor que el de los niños mayores. El factor pronóstico más importante es la NMA. Schmidt et al25 encontraron una supervivencia libre de episodios (SLE) del 10% para los lactantes en estadio 4 con NMA. Para los no amplificados, el tipo de metástasis también influye en la evolución, y el pronóstico es peor en caso de metástasis óseas visibles en radiología convencional, pleurales o SNC.
Grupo de riesgo altoEs el más numeroso, compuesto por pacientes mayores de 1 año en estadio 4 y cualquier estadio (excepto el 1) con NMA, independientemente de la edad. El pronóstico de estos pacientes sigue siendo malo, incluso con tratamientos muy agresivos, pero ha mejorado notablemente en los últimos años, pasando de una SLE a los 5 años del 8% en 1985, a un 30% a partir de 1990.
Sólo la NMA confiere un valor pronóstico negativo a los tumores que la presentan. El tratamiento de estos pacientes consta de quimioterapia de inducción (se han descrito multitud de esquemas para este fin, casi todos utilizan ciclofosfamida, vincristina, etopósido y carboplatino o cisplatino, en diferentes dosis y con resultados similares26,27), cirugía28, megaterapia con trasplante autólogo29 y tratamiento de enfermedad residual mínima.
No se ha demostrado que el grado de respuesta a la quimioterapia de inducción influya en la evolución a largo plazo.
Tratamiento de la enfermedad residual mínimaCon el uso de la quimioterapia de inducción, cirugía y megaterapia con trasplante autólogo, la tasa de respuesta oscila entre el 60 y el 80% de todos los pacientes, pero muchos de estos pacientes recaen. Para intentar controlar este fenómeno, se están utilizando agentes diferenciadores, como los retinoides y los anticuerpos monoclonales.
Agentes diferenciadoresEl ácido 13 cisretinoico induce la diferenciación del neuroblastoma y produce apoptosis in vitro30.
Anticuerpos monoclonalesLos gangliósidos son antígenos de superficie de la membrana que están presentes en todas las células del neuroblastoma y el sistema nervioso. Se han desarrollado anticuerpos monoclonales contra ellos31.
Metayodobencil guanidinaEl 123I-MIBG es una técnica usada en el estudio de los pacientes con neuroblastoma para evaluar la extensión de la enfermedad al diagnóstico y la respuesta al tratamiento. El 131I-MIBG, a dosis altas, se ha usado en el tratamiento del neuroblastoma avanzado32, ya que sigue las mismas rutas metabólicas que las catecolaminas y es captado de forma activa por la membrana celular de los tumores de la cresta neural y sus metástasis. Se ha usado también como régimen de acondicionamiento pretrasplante autólogo de progenitores hematopoyéticos, en combinación con quimioterapia. En algunos casos, se utiliza sola o combinada con topotecán o cisplatino para el tratamiento de recaídas o en caso de tumores refractarios.













