



Puntos clave
- •
Los pólipos juveniles son los más frecuentes (80%); comienzan en la primera década de la vida, con localización preferente en el recto.
- •
Los síndromes de poliposis juvenil, Cowden y Peuzt-Jeghers presentan una herencia autosómico dominante.
- •
El síndrome de poliposis juvenil debe sospecharse ante más de 3 pólipos juveniles en colon o cualquier número de pólipos en otra parte del tracto gastrointestinal, también ante cualquier número de pólipos e historia familiar positiva.
- •
La poliposis adenomatosa familiar es un trastorno hereditario autosómico dominante por mutación del gen APC. El 100% de los pacientes desarrolla cáncer de colon si no se tratan.
- •
Existe una poliposis adenomatosa familiar atenuada. Otras variedades asocian tumores: síndrome de Gardner y síndrome de Turcot; la asociada al gen MYH presenta herencia autosómica recesiva.
- •
El cáncer colorrectal hereditario no polipósico es el cáncer hereditario más común. Presenta herencia autosómico dominante. Se debe a mutaciones germinales en los genes MSH2, MLH1, MSH6 y hPMS2. Asocian otros tumores extracolónicos: endometrio, ovario, estómago o vías renales. Lo fundamental es el diagnóstico precoz.
Lectura rápida
La presencia de pólipos en el colon y el recto es común en la infancia, la mayoría son benignos y únicos. Cuando son múltiples, debemos sospechar un síndrome de poliposis intestinal. La mayoría de estos síndromes tienen carácter hereditario y asocian a un aumento del riesgo de cáncer de colon.
Pólipos inflamatorios y hamartomatosos: los pólipos linfoides son formaciones seudopolipoideas, asintomáticas, benignas y autolimitadas. Muy frecuentes en los primeros años de vida. Comparten características con los pólipos inflamatorios, reactivos a la inflamación mucosa por infecciones crónicas. Ambos carecen de potencial neoplásico.
Los pólipos juveniles son los más frecuentes en los niños (80%), suelen ser únicos y no se malignizan. Si existen múltiples pólipos juveniles (50% de los casos) puede tratarse de un síndrome de poliposis juvenil, con herencia autosómico dominante y una incidencia muy baja, 1–1,5 casos/100.000 personas. Se asocia a mutaciones en los genes BMPR1A y SMAD4 en los cromosomas 10 y 18. Tiene riesgo de degeneración maligna.
El síndrome de Bannayan-Riley-Ruvalcaba (B-R-R), causado por la mutación del gen PTEN (cromosoma 10q23.2), aparece como un síndrome de herencia autosómica dominante o como caso esporádico. Asocia múltiples lipomas, macrocefalia, hemangiomas, pólipos intestinales hamartomatosos, retraso del crecimiento y máculas hiperpigmentadas en los genitales masculinos. No se asocian a riesgo de degeneración maligna.
El síndrome de Cowden, con patrón de herencia autosómico dominante, se caracteriza por triquelomas faciales, queratosis acra, pápulas o papilomas, hamartomas benignos y macrocefalia. Causado por una mutación del gen PTEN (cromosoma 10q23.2) con las mismas mutaciones que en el síndrome de B-R-R, por lo que se trata de una expresión fenotípica diferente.
El síndrome de Peuzt-Jeghers: poliposis hamartomatosa de herencia autosómica dominante por mutación en el gen supresor tumoral LKB1/STK11. Las manchas hiperpigmentadas melánicas pardas o azul oscuras típicamente en los labios y la mucosa bucal aparecen alrededor de los 2–3 años y los pólipos se presentan en el periodo de lactante. Presenta riesgo de malignización a largo plazo.
Pólipos adenomatosos: la poliposis adenomatosa familiar es un trastorno hereditario autosómico dominante con multitud de pólipos adenomatosos premalignos en el colon. Se manifiesta en la segunda década de la vida y la malignización en la tercera década. El gen mutado es el APC, con penetrancia variable. Los niños afectados de poliposis adenomatosa familiar (PAF) tienen el 100% de posibilidades de desarrollar cáncer de colon si no son tratados. Pueden presentar otros tumores: cáncer de tiroides y páncreas. Se diagnostican mediante test genéticos y la endoscopia. El tratamiento es exclusivamente quirúrgico anticipándose a la aparición de la degeneración maligna. Existe una forma atenuada con menos de 100 pólipos, predominio en colon derecho y planos, que desarrollan los carcinomas en edades más avanzadas.
La poliposis asociada a gen MYH, variante de PAF, con herencia autosómica recesiva, causada por mutaciones del gen MYH. Presenta adenomas colónicos (entre 5 y 100) en colon derecho y mayor riesgo de cáncer de colon a edades más tardías.
El síndrome de Gardner, otra variedad de PAF, asocia tumores óseos y del tejido blando, que aparecen antes de la poliposis y se diagnostica en la primera década de la vida.
El síndrome de Turcot asocia a la PAF tumores cerebrales malignos; algunos autores lo consideran como una variante del cáncer colorrectal hereditario no polipósico.
Síndromes de etiología incierta: el síndrome de Cronkhite-Canada es una poliposis hamartomatosa difusa no hereditaria que asocia alopecia, distrofia ungueal e hiperpigmentación cutánea. Aunque es una poliposis no maligna, tiene mal pronóstico dada su mortalidad del 60% por las complicaciones.
El síndrome de poliposis hiperplásica es infrecuente y los pacientes tienen incrementado su riesgo de desarrollar carcinoma colorrectal, se recomienda seguimiento endoscópico.
El cáncer colorrectal hereditario no polipósico o síndrome de Lynch es el cáncer colorrectal hereditario más común, responsable del 2–7% de todos los cánceres de colon. Herencia autosómica dominante de alta penetrancia. Se presenta a edades más tempranas que los otros cánceres de colon. Se debe a mutaciones germinales en los genes MSH2, MLH1, MSH6 y hPMS2. Asocia otros tumores: endometrio, ovario, estomago y vías renales. Es fundamental el diagnóstico precoz. Establecido el diagnóstico, la colectomía subtotal con anastomosis ileorrectal y rectoscopias de control, es el tratamiento de elección.
Los pólipos se caracterizan por ser masas delimitadas de tejido que protruyen hacia la luz intestinal. Pueden presentar tamaños y aspectos diferentes, según sean: pediculados, sésiles, únicos o múltiples.
La presencia de sangre roja, fresca, brillante, sin alteraciones del ritmo intestinal y heces de consistencia normal suelen ser su forma más habitual de presentación; también pueden ser causa de prolapso a través del ano, diarrea, invaginación intestinal, obstrucción y dolor abdominal recurrente. En otros casos, no producen ningún síntoma y son descubiertos de forma incidental durante una endoscopia.
La presencia de pólipos en el colon y el recto es una afección común durante la infancia; en la mayoría de los casos, son benignos y únicos. Habitualmente, se trata de pólipos juveniles, que representan alrededor del 80% de los casos; esta denominación juvenil se refiere a la histología de las lesiones y no a la edad de presentación de los mismos.
Cuando son múltiples, debemos sospechar la existencia de un síndrome de poliposis intestinal, cuadros caracterizados por la presencia de múltiples pólipos a lo largo del tubo digestivo. En la mayoría de los síndromes, existe un carácter hereditario y se asocian a un aumento del riesgo de presentar cáncer de colon. A este grupo pertenecen la poliposis adenomatosa familiar y los pólipos del síndrome de Peutz-Jeghers (P-J), que representan menos del 5% de todos los pólipos de la infancia y adolescencia.
La exploración digital del recto, el enema de bario, la ecografía y otros estudios de imagen más sofisticados y modernos se usan para confirmar el diagnóstico antes de la colonoscopia, que se considera la técnica más efectiva para el diagnóstico y el tratamiento de los pólipos colorrectales1.
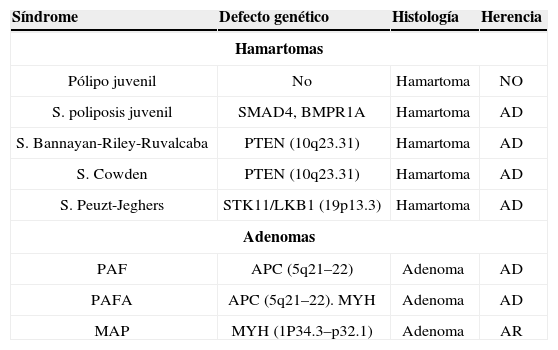
ClasificaciónExisten diversos tipos de clasificaciones, según el número de pólipos, su histología, genética, momento de presentación, etc. Para esta revisión los hemos dividido en una clasificación ecléctica, que nos permitirá repasar la mayoría de los pólipos y síndromes de poliposis en pediatría (tablas 1–3).
Clasificación de pólipos en la infancia
| 1. Inflamatorios y hamartomatosos |
| Pólipos linfoides |
| Pólipos inflamatorios |
| Pólipos juveniles |
| Síndrome de poliposis juvenil |
| Síndrome Bannayan-Riley-Ruvalcaba |
| Síndrome de Cowden |
| Síndrome de Peuzt-Jeghers |
| 2. Adenomatosos |
| Poliposis adenomatosa familiar |
| Poliposis adenomatosa familiar atenuada |
| Poliposis asociada a gen MYH |
| Síndrome de Gardner |
| Síndrome de Turcot tipo 2 |
| 3. Síndromes de etiología incierta |
| Síndrome de Cronkhite-Canada |
| Síndrome de poliposis hiperplásica |
Pólipos en la infancia
| Síndrome | Defecto genético | Histología | Herencia |
| Hamartomas | |||
| Pólipo juvenil | No | Hamartoma | NO |
| S. poliposis juvenil | SMAD4, BMPR1A | Hamartoma | AD |
| S. Bannayan-Riley-Ruvalcaba | PTEN (10q23.31) | Hamartoma | AD |
| S. Cowden | PTEN (10q23.31) | Hamartoma | AD |
| S. Peuzt-Jeghers | STK11/LKB1 (19p13.3) | Hamartoma | AD |
| Adenomas | |||
| PAF | APC (5q21–22) | Adenoma | AD |
| PAFA | APC (5q21–22). MYH | Adenoma | AD |
| MAP | MYH (1P34.3–p32.1) | Adenoma | AR |
MAP: PAP: poliposis adenomatosa familiar; PAFA: poliposis adenomatosa familiar atenuada.
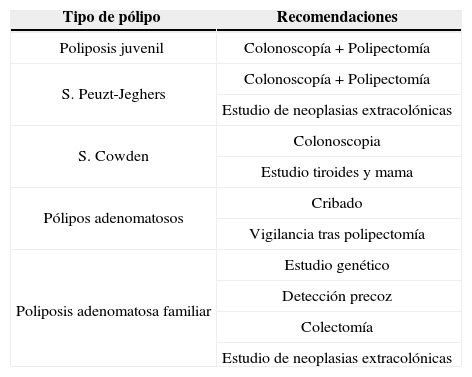
Recomendaciones a seguir en los pólipos intestinales
| Tipo de pólipo | Recomendaciones |
| Poliposis juvenil | Colonoscopía + Polipectomía |
| S. Peuzt-Jeghers | Colonoscopía + Polipectomía |
| Estudio de neoplasias extracolónicas | |
| S. Cowden | Colonoscopia |
| Estudio tiroides y mama | |
| Pólipos adenomatosos | Cribado |
| Vigilancia tras polipectomía | |
| Poliposis adenomatosa familiar | Estudio genético |
| Detección precoz | |
| Colectomía | |
| Estudio de neoplasias extracolónicas |
El primer grupo está formado por los pólipos inflamatorios y aquellos pólipos con apariencia microscópica de hamartomas, generalmente de aspecto no neoplásico y benignos, en los que con poca frecuencia pueden existir trasformaciones neoplásicas o presentar signos histológicos de displasia. Revisaremos los pólipos juveniles, el síndrome de poliposis juvenil, el pólipo linfoide, el inflamatorio, el síndrome de Bannayan-Riley-Ruvalcaba (B-R-R), síndrome de Cowden y el síndrome P-J.
Un segundo tipo son los pólipos adenomatosos. Se trata por definición de lesiones displásicas y precancerosas que presentan facilidad para transformarse en adenocarcinomas. Clásicamente, se dividían en adenoma tubular, tubulovelloso y velloso, pero actualmente tienden a clasificarse según su grado de displasia como de alto y bajo grado2.
Los pólipos de este grupo están generalmente asociados a síndromes de poliposis, tales como la poliposis adenomatosa familiar (PAF), poliposis adenomatosa familiar atenuada (PAFA), poliposis asociada a gen MYH, el síndrome de Turcot y el síndrome de Gardner, que se consideran formas de presentación de la PAF.
El tercer grupo, que no sigue criterios histológicos, está integrado por unas entidades clínicas de etiología incierta y que ocasionalmente pudieran incluirse, no sin dificultades en los grupos anteriores. Revisaremos aquí el síndrome de Cronkhite-Canada, poliposis gastrointestinal no adenomatosa, ni hereditaria, asociada a alteraciones ectodérmicas, y el síndrome de poliposis hiperplásica (SPH).
Pólipos inflamatorios y hamartomatososPólipos linfoidesSon formaciones seudopolipoideas, secundarias a hiperplasia del tejido linfoide intramural, en respuesta a infecciones inespecíficas, víricas generalmente. Su incidencia es alta, aunque, como en la mayoría de los casos, son asintomáticos no llegan a ser diagnosticados. Pueden aparecer en edades muy tempranas de la vida, incluso durante el primer año de edad, aunque lo más característico es que se presenten antes de los 5 años: después de esta edad su presentación es inusual.
Tienen tamaño variable desde pocos milímetros a 3cm de diámetro y son generalmente sésiles en forma. Como su origen es la hiperplasia de los folículos linfoides submucosos y la mucosa que los cubre se ulcera, adquieren un aspecto característico «en volcán» o umbilicados.
Pueden ser causa de anemia por pérdidas ocultas de sangre y, en ocasiones, de sangrado rectal. En algunos casos, puede ser también causa de invaginación intestinal si la afectación es ileocecal o de prolapso rectal si la afectación es distal. Son diagnosticados mediante la colonoscopia al visualizar pequeñas excrecencias uniformes y blanquecinas, que pueden ocasionalmente estar ulceradas en su ápice. El enema opaco con contraste y aire muestra una nodularidad difusa, con nódulos menores de 4 mm, distribuidos de forma regular y rodeados de mucosa normal.
Estos pólipos linfoides son pólipos benignos, autolimitados y con tendencia a la regresión espontánea, carecen por tanto de potencial maligno. Su tratamiento debe ser expectante, con medidas de control, obligando al tratamiento electivo solamente en caso de complicaciones: invaginación, hemorragia, etc.
Pólipos inflamatoriosSon formaciones polipoideas que se desarrollan en respuesta a un proceso inflamatorio crónico de la mucosa intestinal. Son secundarios o reactivos a la inflamación producida por infecciones crónicas: víricas, bacterianas y por la presencia de parásitos. Aparecen en la fase de regeneración y curación de la mucosa. Tienen tamaños variables y suelen ser múltiples. En ocasiones son grandes, presentándose entonces como masas solitarias que impresionan de tumorales. También se presentan como puentes mucosos de un lado al otro de la pared colónica. En la histología aparecen islotes de mucosa que se sobreelevan sobre las zonas circundantes, que habitualmente están ulceradas. No tienen potencial neoplásico intrínseco.
En los niños con enfermedad inflamatoria intestinal, son muy característicos y en la histología presentan, además, las características histológicas de la enfermedad de base (colitis ulcerosa o enfermedad de Crohn). Habitualmente, nos referimos a ellos como seudopólipos.
Pólipos juvenilesLos pólipos más frecuentes en la edad pediátrica son los pólipos juveniles. Se presentan mayoritariamente en niños menores de 10 años, estando el pico de edad de mayor incidencia entre los 2 y 5 años. Aproximadamente, el 50% de los niños presentan más de un pólipo; debemos de estar alerta cuando tengamos un paciente con más de 3–4 pólipos, ya que, en lugar de presentar un pólipo juvenil múltiple, puede ser portador de un síndrome de poliposis juvenil que veremos posteriormente. Macroscópicamente, son pólipos de entre 1 y 3cm, con localización preferente en el colon izquierdo. El 90% de ellos son pedunculados y sus características histológicas típicas son: arquitectura quística, glándulas tortuosas, rellenas de moco con lámina propia prominente e infiltración inflamatoria3. Su presentación clínica habitual es un sangrado rectal indoloro. Otras formas de presentación son el prolapso rectal del pólipo o heces con moco sanguinolento. Su tratamiento de basa en la colonoscopia y la polipectomía. No tienen asociado un riesgo de presentar cáncer; sin embargo, es difícil establecer el punto de corte para clasificar cuando un paciente entra dentro del fenotipo de síndrome de poliposis juvenil.
Síndrome de poliposis juvenilEl síndrome de poliposis juvenil es un síndrome de herencia autosómico dominante. Tiene una incidencia muy baja, entre 1 y 1,5 casos cada 100.000 personas, con 2 formas posibles de presentación, la esporádica y la familiar, ambas con un patrón hereditario autosómico dominante.
Las características que se deben de reunir para establecer su diagnóstico son3–5:
- –
Más de 5 pólipos juveniles colorrectales.
- –
Pólipos juveniles en el tracto gastrointestinal (localización ajena al colon).
- –
Historia familiar de poliposis juvenil.
Su diagnóstico se suele realizar entre el final de la edad infantil y el comienzo de la adolescencia. La manera de presentación más habitual es la del sangrado rectal; otras más infrecuentes son el retraso de crecimiento, la anemia, la hipoalbuminemia o el dolor abdominal. Según su presentación clínica, el síndrome de poliposis juvenil, se puede dividir en 3 grupos fenotípicos: poliposis juvenil de la infancia, poliposis juvenil colónica (solo afecta al colon) y poliposis juvenil generalizada.
La más grave y la de peor pronóstico es el fenotipo de poliposis juvenil de la infancia, con una presentación temprana con diarrea sanguinolenta, enteropatía pierde proteínas, anemia, edemas, y complicaciones derivadas, como la invaginación intestinal. En cambio, las otras 2 formas, poliposis juvenil colónica y la poliposis juvenil generalizada, suelen manifestarse en la primera y la segunda décadas de la vida.
Esta poliposis se caracteriza histopatológicamente por la existencia de numerosísimos pólipos (50–200), de tamaño variable (pocos milímetros a pocos centímetros), rodeados de mucosa normal y distribuidos por todo el colon, a veces incluso en estómago e intestino delgado.
Se asocia a mutaciones genéticas del gen SMAD4, localizado en el cromosoma 18, y el BMPR1A, localizado en el cromosoma 10. Este síndrome se caracteriza por el riesgo de degeneración maligna. Es importante realizar el diagnóstico molecular para poder ofrecer al paciente un pronóstico con respecto al riesgo de desarrollar cáncer.
Los niños con un número de pólipos limitado pueden ser tratados con polipectomía endoscópica. Si se detecta un alto grado de displasia y no puede resecarse completamente, debe practicarse una colectomía (tabla 3).
Cuando el número de pólipos es muy alto, no corresponde tratamiento endoscópico y es mandataria la colectomía total con anastomosis ileorrectal, o una proctocolectomía total con anastomosis ileoanal, siempre con seguimiento del segmento colónico residual.
En los casos con pólipos del tramo alto, debe intentarse el tratamiento endoscópico y, si no es posible, recurrir a la cirugía.
El seguimiento en estos pacientes se realizará de la siguiente manera6:
- –
En pacientes sintomáticos: colonoscopia cada 3 años desde el comienzo clínico.
- –
En pacientes con antecedentes familiares se realizará colonoscopia bianual desde los 15 años.
El síndrome de B-R-R es un síndrome raro causado por la mutación del gen PTEN, localizado en el cromosoma 10q23.2. Puede presentarse como un síndrome de herencia autosómica dominante o también como casos esporádicos7. Se caracteriza por la asociación de múltiples lipomas, macrocefalia, hemangiomas, pólipos intestinales hamartomatosos, retraso del crecimiento y máculas hiperpigmentadas en los genitales masculinos.
Entre el 35–45% de los casos, presentan pólipos intestinales. No se asocian a riesgo de degeneración maligna. Se pueden localizar en cualquier tramo del tracto gastrointestinal, aunque son más frecuentes en el colon y el recto. Durante la infancia, su forma de presentación es diarrea, dolor abdominal, sangre en heces, anemia, invaginación u obstrucción intestinal. El seguimiento de estos pacientes será la detección anual de sangre oculta en heces y control de hemoglobina7.
Síndrome de CowdenEl síndrome de Cowden, también conocido como síndrome de hamartomas múltiples, es una enfermedad de origen genético que se transmite según un patrón de herencia autosómico dominante y se caracteriza por triquelomas faciales, queratosis acra, pápulas o papilomas, hamartomas benignos y macrocefalia. Se presenta de forma más frecuente en mujeres de raza caucásica. Se debe a una mutación del gen PTEN, localizado en el cromosoma 10q23.2; de hecho, se han encontrado las mismas mutaciones que en el síndrome de B-R-R, por lo que se consideran 2 formas de diferente expresión fenotípica8.
Se asocia a cáncer de mama, tiroides, endometrio, adenocarcinoma colorrectal, linfoma no Hodking y melanoma, entre otros.
Las últimas publicaciones recomiendan para el seguimiento de estos pacientes:
- 1.
En menores de 18 años, ecografía tiroidea, examen dermatológico, neurológico y psicológico.
- 2.
En mayores de 18 años: ecografía anual tiroidea, mamografía anual, biopsia endometrial, colonoscopia y ecografía renal bianual.
Se trata de una poliposis hamartomatosa de herencia autosómica dominante. La mutación se encuentra en el gen supresor tumoral LKB1/STK119,10. El signo que debe poner en alerta al clínico para su diagnóstico es la aparición de manchas hiperpigmentadas melánicas pardas o azul oscuras típicamente en los labios y la mucosa bucal, aunque también se pueden presentar en la cara, manos, pies, encías o paladar. La edad de aparición de estas manchas suele ser alrededor de los 2–3 años, los pólipos se presentan en el periodo de lactante. La mitad de los pacientes afectados de este síndrome presentarán manifestaciones clínicas antes de los 20 años.
Los pólipos son múltiples y lobulados, de superficie lisa, y pueden afectar a todo el tracto intestinal, desde el esófago hasta el recto.
El cuadro clínico se corresponde con un paciente anémico con cuadros de invaginación recidivante. Pueden encontrarse pólipos también en localización extraintestinal, como en los uréteres, vejiga y riñón. En las mujeres, este síndrome se asocia a tumores de ovario.
Debido a que existe un riesgo de malignización a largo plazo, el seguimiento debe realizarse mediante ecografía de tiroides, ecografía pélvica en las mujeres y testicular en los varones.
El tratamiento depende de la clínica, el número de pólipos y su localización. Los niños con pólipos solamente presentes en el intestino delgado y asintomáticos no precisan tratamiento. Si los pólipos se encuentran agrupados en una zona concreta, se realizará una resección, siendo lo más económica posible. En aquellos casos en los que los pólipos sean gastroduodenales y en el recto, será necesario realizar un seguimiento mediante endoscopias anuales con biopsia para su control.
Pólipos adenomatososPoliposis adenomatosa familiar (PAF)La PAF familiar es un trastorno hereditario autosómico dominante en el que se desarrollan multitud de pólipos adenomatosos premalignos en el colon.
Las manifestaciones clínicas se suelen presentar alrededor de la segunda década de la vida y la malignización suele ocurrir en la tercera década. Existen pacientes con malignización más temprana y se corresponden a casos homocigotos para el gen causante de la poliposis.
El gen en el que se produce la mutación es el APC (adenomatous polyposis coli), localizado en el cromosoma 5q21, con una penetrancia variable11. La función de este gen es crucial en diferentes procesos celulares, como es la adhesión y la migración celular. Una alteración en su regulación sería la causante del inicio y la expansión del cáncer de colon. A pesar de esto, un tercio de los pacientes afectados de PAF presentan mutaciones de novo.
Los pacientes afectados de PAF tienen el 100% de posibilidades de desarrollar cáncer de colon si no son tratados.
La sintomatología que presentan está en relación directa con el número de pólipos. Presentan además un riesgo elevado para desarrollar otros tumores extraintestinales como el cáncer de tiroides y páncreas.
El diagnóstico se realizará mediante test genéticos, para estudiar la presencia de mutaciones, y la endoscopia11, en la que se suelen visualizar unos pólipos de aspecto aframbuesado.
- –
En pacientes con historia familiar pero en los que no se ha demostrado alteración genética, se realizará colonoscopia anual desde los 12 años hasta los 30 años. A partir de los 30 años, si no ha existido lesión, se podrá espaciar la colonoscopia cada 3-5 años.
- –
En los pacientes con mutación conocida, se realizará colonoscopia anual desde los 10 años.
El tratamiento de esta entidad es exclusivamente quirúrgico, anticipándose a la aparición de la degeneración maligna. Existen controversias a la hora de establecer tanto la edad de tratamiento como la técnica quirúrgica.
En cuanto a la edad para realizar la intervención, la mayoría de los trabajos consultados la fijan en los 25 años.
Con respecto a la técnica quirúrgica, la más extendida es la colectomía asociada a mucosectomía rectal con reservorio ileal. Los controles postoperatorios deben centrarse en la mucosa para valorar cualquier aparición de pólipos y proceder a su extirpación.
Estudios acerca de la quimioprevención indicaron que el empleo de antiinflamatorios no esteroideos disminuían el número de pólipos, pero este efecto era tan solo pasajero, ya que la supresión de la medicación se acompañaba de una reaparición de las lesiones12.
Poliposis adenomatosa familiar atenuadaEs una forma menos expresiva de la PAF, en la que el número de pólipos es menor a 100, predominantes en el colon derecho y habitualmente planos3. El desarrollo de carcinomas en esta forma se presenta en edades más avanzadas.
Poliposis asociada a gen MYHEs una variante de la PAF que se hereda de forma autosómica recesiva y se produce por mutaciones del gen MYH.
Se caracteriza por adenomas colónicos (entre 5 y 100), predominantes en el colon derecho, y presentan también un riesgo mayor de presentar cáncer de colon, aunque a edades más tardías13–15.
Síndrome de GardnerEl síndrome de Gardner es una variedad de PAF que se asocia a tumores óseos y del tejido blando16.
La mayoría de estos pacientes son diagnosticados en la primera década de la vida. Los tumores óseos y cutáneos aparecen antes de la poliposis.
El 70% de estos pacientes presentan alteraciones dentales asociadas, por lo que en un paciente con osteomas y antecedentes familiares de poliposis colónica habrá que descartar el síndrome de Gardner. Los pólipos suelen afectar principalmente al colon derecho, aunque se ha descrito hasta en un 12% de casos pólipos gástricos y en intestino delgado.
Síndrome de TurcotEs la asociación de PAF y tumores cerebrales malignos. El tipo I es de herencia autosómica recesiva y se debe a una alteración en el gen MLH1 localizado en el brazo corto del cromosoma 3. El tipo II es autosómico dominante y es el resultado de una mutación del gen APC17. Ante un paciente con PAF y síntomas neurológicos, es obligado descartar un proceso invasivo cerebral.
Es importante destacar que tanto la PAFA como poliposis asociada al gen MYH y el síndrome de Turcot son variantes de la PAF en las que se debe de hacer un cribado de cáncer colon, así como de otros tumores extraintestinales. Otros autores consideran el síndrome de Turcot como una variante del síndrome de Lynch o cáncer colorrectal hereditario no polipósico.
Síndromes de etiología inciertaSíndrome de Cronkhite-CanadaEs un poliposis gastrointestinal hamartomatosa difusa, no hereditaria, en la que se asocian alopecia, distrofia ungueal e hiperpigmentación cutánea.
Su etiopatogenia no está aclarada. Suele iniciarse en la sexta década y presenta una mayor predilección por el sexo masculino.
A pesar de ser considerada una poliposis no maligna, ya que los pólipos son hamartomatosos, el pronóstico en general es malo. Estos pacientes presentan importantes cuadros de desnutrición debido a la diarrea que asocian, crisis de invaginación, perforación colónica, con una mortalidad del 60% en su curso evolutivo18. Por ser una enfermedad de afectación difusa, el tratamiento quirúrgico no está indicado y el tratamiento médico es paliativo. Algunos casos se han beneficiado del tratamiento combinado con corticoides, cromoglicato sódico, ranitidina, loratadina y ciprofloxacino. A pesar de no ser considerado una poliposis maligna, existen publicaciones sobre su degeneración a adenocarcinomas gastrointestinales.
Síndrome de poliposis hiperplásicaCuadro infrecuente que se caracteriza fenotípicamente por la presencia de pólipos hiperplásicos múltiples, incluyendo algunos de gran tamaño y localizados en el colon proximal, se asocia ocasionalmente a adenomas serrados o pólipos mixtos. El diagnóstico se realiza basándose en criterios básicamente clínicos19:
- a.
Cinco o más pólipos hiperplásicos proximales al colon sigmoide, y al menos 2 de ellos mayores de 10 mm.
- b.
Cualquier número de pólipos hiperplásicos proximales al colon sigmoide en pacientes que tengan un familiar de primer grado con poliposis hiperplásica.
- c.
Más de 30 pólipos hiperplásicos de cualquier tamaño pero distribuidos en todo el colon.
El SPH posee gran variabilidad de presentación en relación con el número y el tamaño de los pólipos, la edad de presentación y la asociación a cáncer colorrectal, y la historia familiar de carcinoma colorrectal. En el momento actual, se desconoce su base genética20.
Inicialmente, se consideró una entidad benigna, pero se ha descrito una incidencia variable de carcinoma colorrectal, por lo que se considera que los pacientes que cumplen los criterios clínicos señalados tienen incrementado su riesgo de desarrollar carcinoma colorrectal.
La anatomía patología se caracteriza por pólipos con arquitectura serrada en la porción proximal de las criptas o bien con la existencia en la base de las criptas de dilatación y proliferación displásica. Otros casos son de tipo mixto: componente hiperplásico asociado a adenomatoso.
Existen pólipos de histología hiperplásica, que no cumplen los criterios de este síndrome. Son pólipos no neoplásicos, normalmente pequeños (< 5mm), en escaso número y distribuidos en el recto-sigma.
Comentario final. Síndrome de Lynch o cáncer colorrectal hereditario no polipósicoNo debemos finalizar esta revisión sin mencionar cáncer colorrectal hereditario no polipósico (CCHNP) o síndrome de Lynch, ya que es el cáncer colorrectal hereditario más común y además el responsable de entre el 2 y el 7% de todos los cánceres de colon21.
El CCHNP se presenta a edades más tempranas que los otros cánceres de colon. Existen numerosos casos de CCHNP de aparición en la edad pediátrica, el más pequeño de 6 años de edad, de ahí la razón para este comentario final22–24.
El CCHNP es un proceso con herencia autosómica dominante y una alta penetrancia. La predisposición genética a padecer CCHNP se debe a las mutaciones germinales en los genes MSH2, MLH1, MSH6 y hPMS2 que codifican las proteínas del sistema de reparación del ADN (mismatch repair system), lo que va a producir daños en los cromosomas con fragmentación y formación de microsatélites, lo que se conoce como inestabilidad de microsatélites (MSI), precisamente el estudio de esta inestabilidad o MSI será fundamental para el diagnóstico de estos pacientes25.
El CCHNP se presenta a una edad entre 45–50 años, con tumores en colon proximal, pudiendo presentar otros tumores extracolónicos: endometrio, ovario, estómago o vías renales. Según el tipo de mutación, existen unos riesgos diferenciados; así, los portadores de mutaciones de MSH2 son los que presentan un mayor riesgo cáncer a lo largo de su vida y asocian tumores uroteliales con más frecuencia. Por el contrario, los portadores de mutación de MSH6 presentan mayor riesgo de cáncer de endometrio y un riesgo menor y a edad más tardía de presentar cáncer de colon.
Para identificar a aquellos pacientes de riesgo, se utilizan los criterios de Ámsterdam II y también las guías de Bethesda, que tratan de identificar a pacientes subsidiarios de análisis de MSI26.
La actitud primordial es el diagnóstico precoz, realizando colonoscopias anuales desde los 20–25 años, sin olvidar el cribado anual de cáncer de endometrio, así como gastroscopia y citologías de orina para investigar los posibles tumores gástricos y uroteliales27.
Cuando se establece el diagnóstico, la colectomía subtotal con anastomosis ileorrectal y rectoscopias de control, es el tratamiento de elección, ya que la mayoría de las guías no recomiendan la quimioterapia paliativa.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.