








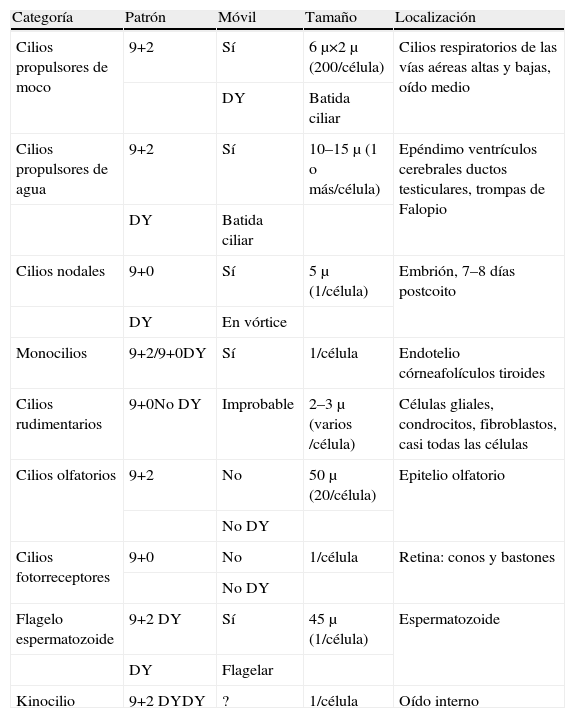
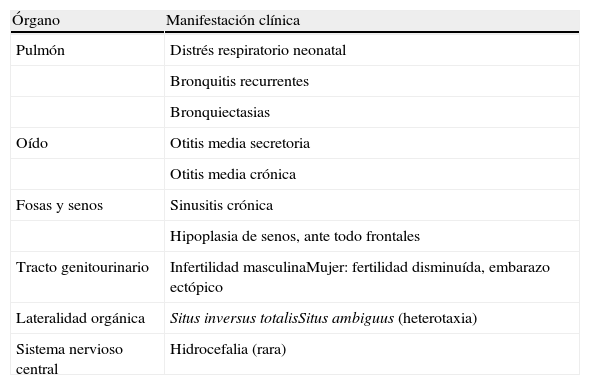

La discinesia ciliar primaria es un trastorno genéticamente determinado que se caracteriza por un movimiento ciliar alterado o ausente. Genera un déficit en el aclaramiento mucociliar que se manifiesta clínicamente como infecciones crónicas de vías aéreas constantes desde el nacimiento, así como esterilidad masculina por inmovilidad del espermatozoide y situs inversus en el 40–50% de los pacientes (síndrome de Kartagener). El diagnóstico se basa en el estudio de la movilidad ciliar mediante vídeo de alta resolución digital y alta velocidad, complementado con el estudio de la ultraestructura ciliar. La amplia distribución ciliar en el organismo y sus numerosas funciones hacen que su patología origine, además de la discinesia ciliar primaria, otras ciliopatías.
Primary ciliary dyskinesia is a genetically inherited syndrome characterized by cilia immotility or dysmotility. Deficiency in mucociliary clearance produces chronic respiratory infections since birth, male sterility by spermatozoid immotility and situs inversus in 40–50% of patients (Kartagener's syndrome). Diagnosis is made by analyzing cilia motility with high-speed digital video and ciliar ultrastructure. The wide distribution and functions of the cilia in the body mean that this dysfunction can generate other ciliopathies apart from primary ciliary dyskinesia.