






Cardiac diseases are responsible for 80% of sudden deaths, but they may also be involved in deaths that have occurred during surgical procedures or in hospitalised patients, as well as being related to accidental deaths. A specialised heart examination guarantees the medico-legal assessment of the cause of death, and is essential in the diagnoses of familial cardiopathies. The pathological characteristics, according to cardiovascular pathology criteria of the most frequent structural cardiopathies in forensic practice are presented.
La patología cardiaca no solo es responsable del 80% de las muertes súbitas, sino que puede ser coadyuvante en las muertes ocurridas durante intervenciones quirúrgicas o en el transcurso de ingresos hospitalarios, y puede estar relacionada con muertes accidentales. Un estudio especializado y protocolizado del corazón garantiza una correcta valoración médico-legal del fallecimiento, pero además es esencial para la detección de cardiopatías familiares. Se presentan las características anatomopatológicas de las cardiopatías estructurales más frecuentes en el ámbito forense según criterios de los grupos de referencia en patología cardiovascular.
Heart disease is not only responsible for 80% of sudden deaths (SDs), but may also be involved in deaths that have occurred during surgical procedures or hospital admissions, and may be related to road traffic accidents, or accidents which have occurred at work or at home. Likewise, death may be as a result of complications associated with interventions on the cardiovascular system, such as corrections of congenital heart disease, coronary intervention, ablations, valve replacement, pacemaker placement, etc.1 Due to all of these circumstances, the histopathological study of the heart is the most requested study in the judicial field. A specialised and standardised study of the heart guarantees the correct medico-legal assessment of the death, but it is also essential for the detection of inherited heart diseases.
Cardiovascular autopsyThere are several publications concerning cardiovascular autopsy.1,2 One of the most important publications is that issued by the Association for European Cardiovascular Pathology,3 the update of which will be published in the next few weeks. The diagnostic criteria of the different heart diseases, such as cause of death, which is extremely important regarding implications for family members, are listed in these guidelines and other studies.4
Although it is not always accessible to the medical examiner, clinical information is essential to appropriately guide the heart study: personal cardiac history (syncopal episodes, chest pain, HTN, implantation of coronary stents or valve replacements, etc.) and extracardiac history, as well as family history of heart disease. Due to the fact that numerous heart diseases are inherited, the sampling of blood (in EDTA) and of fresh tissue is essential for genetic studies.3
The study of the heart starts with the opening up of the rib cage to rule out haemothorax or haemomediastinum which may be secondary to rupture of the thoracic aorta. The contents of the pericardial sac, both its volume and characteristics, are then studied. Haemopericardium should make us consider a rupture of the ascending aorta or ruptured acute myocardial infarction (AMI). With the heart in situ, the pulmonary trunk should be sectioned looking for thrombi which may have lodged into both pulmonary arteries. The heart is completely dissected with both atria intact. Normal heart weight is not an absolute value but, rather, depends on body weight and height. Reference tables therefore have to be used.1,5 It is advisable to perform an X-ray of the entire heart to locate coronary stents and assess prostheses. Please refer to Morentin et al.2 for a thorough description of the heart study method. We will now move on to discuss the most common diseases.
Coronary artery diseaseThis is the most common heart disease in forensic practice given that 60–90% of SDs are attributed to this disease. We found ischaemic heart disease, predominantly chronic, both of the coronary arteries (with stenosis >75% of the lumen) (92%), and of the myocardium (scars) (39%) in 65% of the SDs studied in our department.6 The chronic stenotic lesions of the coronary arteries are divided into two large groups: stable plaques with thick fibrous capsules on a lipid or fibrous necrotic centre (atheromatous plaques themselves) or pathological intimal hyperplasia (juvenile atherosclerosis).7 The incidence of acute coronary thrombosis is variable (we found it in 27% of cases),6 and it can be caused by the rupture of atheromatous plaques with thin fibrous capsules (vulnerable plaques) which result in the content of the plaque being excreted and intraluminal fibrin-platelet thrombosis (Fig. 1A); or to endothelial denudation of the plaque (erosion) with occlusive or mural platelet thrombosis (Fig. 1B); this type occurs in stable atheromatous plaques or in pathological intimal hyperplasia. Plaque rupture occurs more frequently in males aged 35–80 with classic risk factors (hyperlipidaemia, smoking, diabetes) and erosion occurs more frequently in younger people associated with smoking and genetic factors.7 The link between cocaine use and early atheromatosis with coronary thrombosis is important in young people.8,9
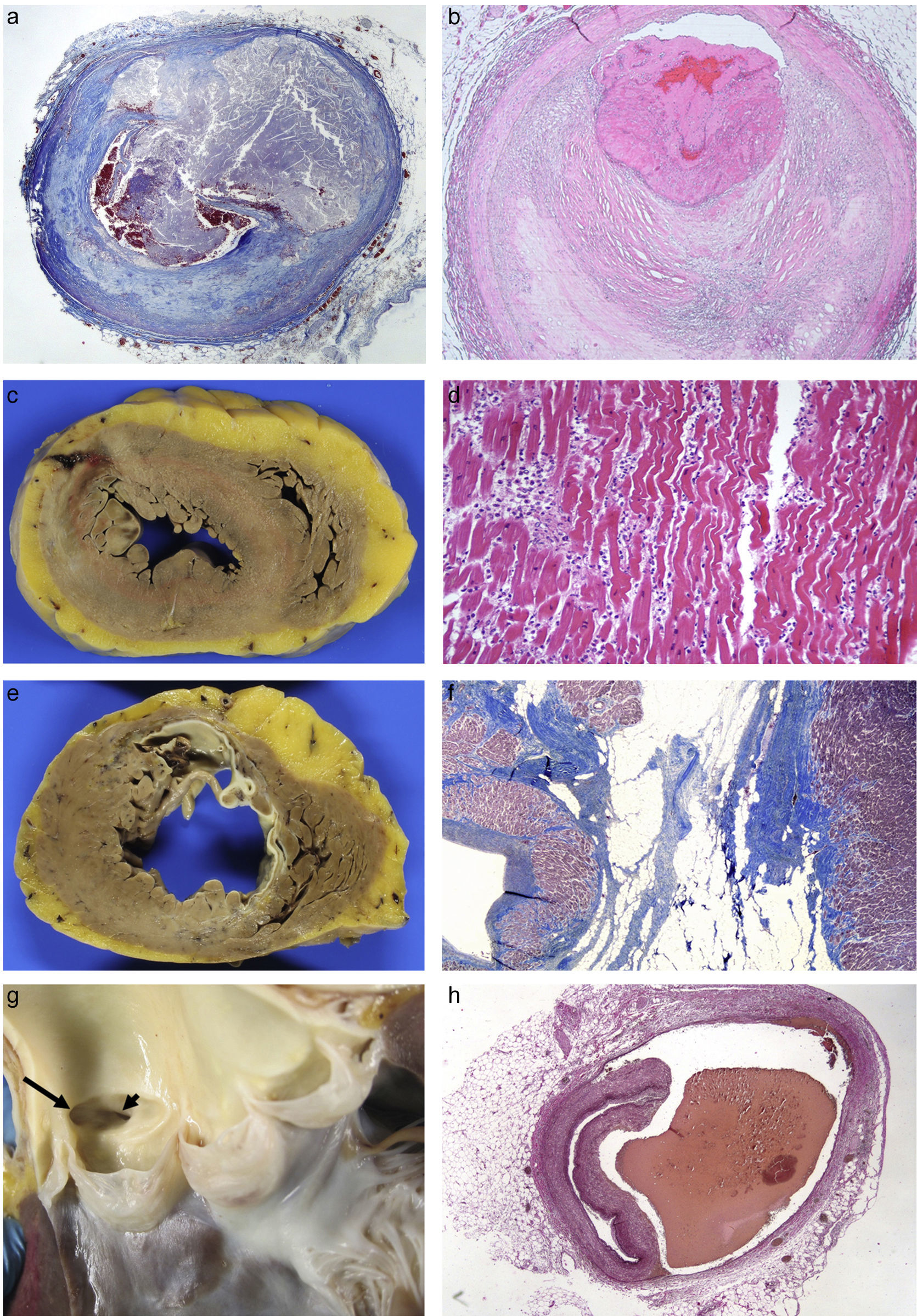 24h: necrosis due to coagulation of the myocytes and intense neutrophilic inflammatory infiltrate (HE, 20×). (E) Previous transmural anteroseptal infarction with marked wall thinning and endocardial fibrosis. (F) Dense collagen scar with extensive adipose metaplasia (Masson's trichrome 4×). (G) Origin of the two coronary arteries in the right sinus (long arrow in left ostium; short arrow in right ostium). (H) Coronary artery dissection with haematoma between the middle layer and the external elastic lamina (Weigert, 2×).' title='(A) Rupture of vulnerable plaque with secretion of content (Masson's trichrome 2×). (B) Plaque erosion and occlusive platelet thrombosis (HE, 4×). (C) Anterior transmural AMI with wall rupture. (D) AMI >24h: necrosis due to coagulation of the myocytes and intense neutrophilic inflammatory infiltrate (HE, 20×). (E) Previous transmural anteroseptal infarction with marked wall thinning and endocardial fibrosis. (F) Dense collagen scar with extensive adipose metaplasia (Masson's trichrome 4×). (G) Origin of the two coronary arteries in the right sinus (long arrow in left ostium; short arrow in right ostium). (H) Coronary artery dissection with haematoma between the middle layer and the external elastic lamina (Weigert, 2×).'/>
24h: necrosis due to coagulation of the myocytes and intense neutrophilic inflammatory infiltrate (HE, 20×). (E) Previous transmural anteroseptal infarction with marked wall thinning and endocardial fibrosis. (F) Dense collagen scar with extensive adipose metaplasia (Masson's trichrome 4×). (G) Origin of the two coronary arteries in the right sinus (long arrow in left ostium; short arrow in right ostium). (H) Coronary artery dissection with haematoma between the middle layer and the external elastic lamina (Weigert, 2×).' title='(A) Rupture of vulnerable plaque with secretion of content (Masson's trichrome 2×). (B) Plaque erosion and occlusive platelet thrombosis (HE, 4×). (C) Anterior transmural AMI with wall rupture. (D) AMI >24h: necrosis due to coagulation of the myocytes and intense neutrophilic inflammatory infiltrate (HE, 20×). (E) Previous transmural anteroseptal infarction with marked wall thinning and endocardial fibrosis. (F) Dense collagen scar with extensive adipose metaplasia (Masson's trichrome 4×). (G) Origin of the two coronary arteries in the right sinus (long arrow in left ostium; short arrow in right ostium). (H) Coronary artery dissection with haematoma between the middle layer and the external elastic lamina (Weigert, 2×).'/>(A) Rupture of vulnerable plaque with secretion of content (Masson's trichrome 2×). (B) Plaque erosion and occlusive platelet thrombosis (HE, 4×). (C) Anterior transmural AMI with wall rupture. (D) AMI >24h: necrosis due to coagulation of the myocytes and intense neutrophilic inflammatory infiltrate (HE, 20×). (E) Previous transmural anteroseptal infarction with marked wall thinning and endocardial fibrosis. (F) Dense collagen scar with extensive adipose metaplasia (Masson's trichrome 4×). (G) Origin of the two coronary arteries in the right sinus (long arrow in left ostium; short arrow in right ostium). (H) Coronary artery dissection with haematoma between the middle layer and the external elastic lamina (Weigert, 2×).
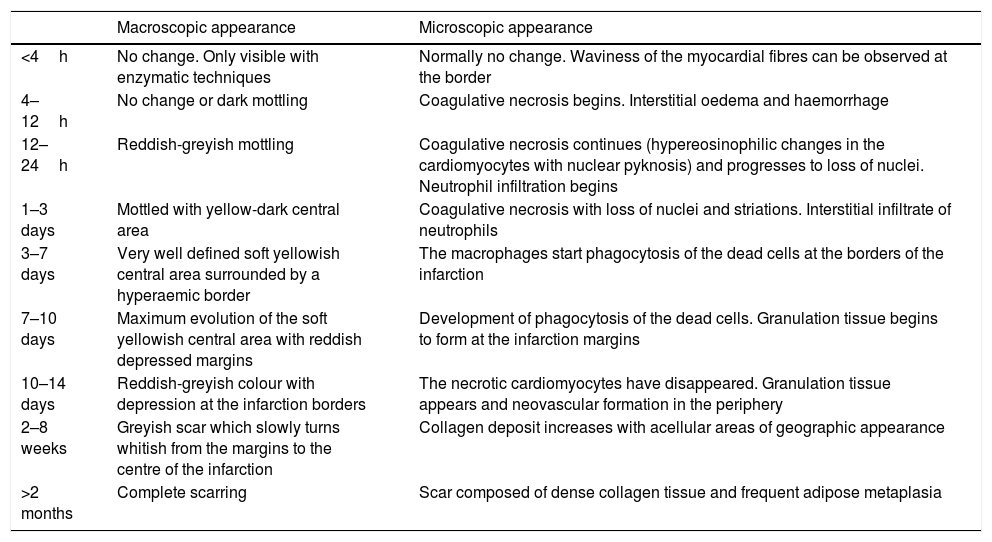
Coronary thrombosis causes AMI but, when death occurs at an early stage (in less than 12h) from the coronary occlusion, there is no time for the ischaemic necrosis in the myocardium to be visible. The dating of the AMI is a very important issue in forensic pathology, because the deceased frequently present with symptoms hours before death for which medical attention is requested. Table 1 summarises the chronology of an infarction adapted from Schoen10 (Fig. 1C–F).
Time course of non-reperfused myocardial infarction.
| Macroscopic appearance | Microscopic appearance | |
|---|---|---|
| <4h | No change. Only visible with enzymatic techniques | Normally no change. Waviness of the myocardial fibres can be observed at the border |
| 4–12h | No change or dark mottling | Coagulative necrosis begins. Interstitial oedema and haemorrhage |
| 12–24h | Reddish-greyish mottling | Coagulative necrosis continues (hypereosinophilic changes in the cardiomyocytes with nuclear pyknosis) and progresses to loss of nuclei. Neutrophil infiltration begins |
| 1–3 days | Mottled with yellow-dark central area | Coagulative necrosis with loss of nuclei and striations. Interstitial infiltrate of neutrophils |
| 3–7 days | Very well defined soft yellowish central area surrounded by a hyperaemic border | The macrophages start phagocytosis of the dead cells at the borders of the infarction |
| 7–10 days | Maximum evolution of the soft yellowish central area with reddish depressed margins | Development of phagocytosis of the dead cells. Granulation tissue begins to form at the infarction margins |
| 10–14 days | Reddish-greyish colour with depression at the infarction borders | The necrotic cardiomyocytes have disappeared. Granulation tissue appears and neovascular formation in the periphery |
| 2–8 weeks | Greyish scar which slowly turns whitish from the margins to the centre of the infarction | Collagen deposit increases with acellular areas of geographic appearance |
| >2 months | Complete scarring | Scar composed of dense collagen tissue and frequent adipose metaplasia |
Congenital abnormalities originating in the coronary arteries account for the second biggest cause of SD in competitive American athletes,11 and for 2–4% of SDs in recreational athletes in our setting.12 Several types can be found: origin of the left coronary artery in the pulmonary trunk or in the right coronary sinus (Fig. 1G); origin of the right coronary artery in the left sinus; or origin of either of the two several mm above the sinotubular junction.13 The origin of the left coronary artery in the pulmonary artery trunk results in the left ventricle (LV) receiving poorly-oxygenated blood, which induces the development of collateral branches from the right coronary artery to compensate for hypoxia but, in situations of greater oxygen demand, such as sporting activity, it can cause SD.6,12 The consequences of the coronary artery originating from the opposite sinus depend on the acute angle of the ostium and its path from its origin to being located in its normal position. The acute angle and the course of the anomalous coronary artery between the aorta and the pulmonary artery, or embedded in the aortic wall, makes them susceptible to collapsing during physical exercise.
The intramyocardial course of the anterior descending coronary artery is a very common finding, reported in 21% of autopsied hearts. It is considered to be of greater importance when it has a length of 2–3cm, a depth of 2–5mm, the surrounding myocardium is arranged in the form of a sphincter around the coronary artery and there is fibrosis.13,14
Spontaneous coronary artery dissection consists of the formation of a haematoma between the middle layer and the external elastic which causes its lumen to collapse (Fig. 1H). It tends to affect the left coronary artery and its branches and its origin may be spontaneous or iatrogenic due to intimal rupture in coronary intervention techniques. Its spontaneous form occurs more frequently in women, related to pregnancy, the postpartum period, fertility treatment and the use of contraceptives. It is also associated with collagenopathies, use of cocaine, smoking, HTN, intense physical exercise, weight lifters or even prolonged spells of sneezing. Eosinophils are often microscopically identified in the adventitia.13
The coronary arteries are also susceptible to vasculitis, which can compromise their lumen and cause SD due to ischaemia. They usually form part of the systemic vasculitides, such as Kawasaki disease, Takayasu disease, giant-cell arteritis, Churg-Strauss syndrome, Wegener's disease or collagenopathies.13,14 Kawasaki disease is more common in children. It tends to progress to the formation of aneurysms which can become thrombosed and cause myocardial ischaemia. Takayasu arteritis affects young adults and causes stenotic lesions of the ostia and of the proximal sections of the coronary arteries, although it can also cause multiple segmental lesions and aneurysms.14
CardiomyopathiesThey include a wide range of diseases in which the heart muscle is functionally and structurally abnormal in the absence of coronary artery disease, hypertensive heart disease or heart valve disease, or in the absence of sufficient congenital heart disease to cause the abnormalities observed.15 According to the American Heart Association, cardiomyopathies are a heterogeneous group of myocardial diseases associated with mechanical and/or electrical dysfunction. They are classified into primary diseases if the disease is confined to the heart, or secondary diseases if the cardiomyopathy forms part of a systemic disease. They are also classified in accordance with their aetiology into genetic, acquired or mixed diseases.16 Genetic cardiomyopathies generally have an autosomal dominant inheritance pattern. It is therefore fundamental to give appropriate advice to first-degree relatives.
SD may manifest in people with no symptoms or with non-specific mild discomfort and therefore with no clinical diagnosis. There is a clear predominance of males in SD series.17 The most common cardiomyopathies in forensic pathology, both in the general population and in athletes, are myocarditis and hypertrophic, arrhythmogenic and dilated cardiomyopathies.
MyocarditisInflammatory disease of the myocardium with various causes: infectious agents, vasculitis, sarcoidosis, drugs, radiation, toxic substances, poisons and hypersensitivity. The clinical spectrum is very varied, with cases that start with SD or that present with mild symptoms, dyspnoea, precordial pain or heart failure.18 We have diagnosed diffuse myocarditis in cases referred as submersion.
Viral infections are the main cause of myocarditis, particularly enterovirus, adenovirus and parvovirus B19.19 The frequency of myocarditis as a cause of SD is highly variable, depending on age and the population studied. Myocarditis causes 4% of sport-related deaths in Spain.12 The diagnosis of myocarditis is microscopic and established histological and immunohistochemical criteria should be followed to determine if it is the cause of SD.3,4,20 Fifty per cent present with a macroscopically normal myocardium (Fig. 2A); sometimes it is mottled and congestive or with whitish areas of fibrosis and the ventricles may be dilated.
 Macroscopically normal heart; (B) Microscopic study finding of myocyte necrosis and lymphohistiocytic infiltrate (HE, 40×). Hypertrophic cardiomyopathy: (C) Asymmetric hypertrophy with fasciculated appearance of the septal myocardium (mutation in the MYBPC3 gene); (D) myocyte hypertrophy, disorder and arterial branches with very thick walls and reduced lumen (HE, 20×). Arrhythmogenic RV cardiomyopathy: (E) Dilated ventricle, with replacement of the myocardium by tissue with a transmural adipose appearance and formation of aneurysms on the anterior, lateral and posterior sides (arrows); (F) Microscopic appearance where adipose wall with fibrous tissue and limited numbers of myocytes along the subendocardial border are observed (Masson")
Acute myocarditis: (A) Macroscopically normal heart; (B) Microscopic study finding of myocyte necrosis and lymphohistiocytic infiltrate (HE, 40×). Hypertrophic cardiomyopathy: (C) Asymmetric hypertrophy with fasciculated appearance of the septal myocardium (mutation in the MYBPC3 gene); (D) myocyte hypertrophy, disorder and arterial branches with very thick walls and reduced lumen (HE, 20×). Arrhythmogenic RV cardiomyopathy: (E) Dilated ventricle, with replacement of the myocardium by tissue with a transmural adipose appearance and formation of aneurysms on the anterior, lateral and posterior sides (arrows); (F) Microscopic appearance where adipose wall with fibrous tissue and limited numbers of myocytes along the subendocardial border are observed (Masson's trichrome, 10×). Dilated cardiomyopathy: (G) LV cavity with a 5cm diameter, mitral valve perimeter measuring 12cm and myocardium of normal appearance; (H) replacement fibrosis with hypertrophic and thinned myocytes found in histological study (HE, 40×).
The microscopic appearance is variable, from cases that present extensive myocyte necrosis (Fig. 2B) to others with more limited necrosis. In both cases, there is oedema and extensive interstitial inflammatory infiltrate with predominance of T lymphocytes, histiocytes and macrophages that engulf necrotic cells. According to the type of cellular infiltrate, cases of myocarditis are classified into lymphocytic, neutrophilic with formation of abscesses, eosinophilic, giant-cell or with granulomas (sarcoidosis). The predominant form is lymphocytic myocarditis, which is believed to be of viral aetiology, although normally the virus is not detected. Bacteria, fungi, parasites or granulomas are identified more rarely. Isolated inflammatory foci are frequently incidentally found in heart autopsies.
Hypertrophic cardiomyopathyThis is an inherited disease which causes myocardial hypertrophy in the absence of conditions which increase afterload. Hypertrophic cardiomyopathy (HCM) is the most common inherited heart disease (1/500 adults), with an autosomal dominant inheritance. There is intrafamilial variation of the phenotype, meaning that individuals with an identical mutation present with diverse clinical and morphological manifestations. HCM causes SD in young people less than 25 years old, who are asymptomatic or with minimal symptoms, generally without a previous clinical diagnosis. SD associated with sporting activity can occur, predominantly in males.11,12,17
Hearts are heavier, generally >500g, with hypertrophy that compromises the LV, with thickened walls, trabeculae and prominent papillary muscles and reduced cavity. Hypertrophy of the interventricular septum, which is between 2cm and 4cm thick, is striking, with asymmetric anteroseptal hypertrophy being the most common form (Fig. 2C). Hypertrophy may be extensive or only affect the base or the middle third of the ventricles. There is a rare form of apical hypertrophic cardiomyopathy. The right ventricle (RV) may be compromised, with hypertrophic wall, trabeculae and moderator band. In the myocardium, there are usually whitish scarred foci of irregular size and distribution or, in cases of a more advanced clinical stage, more diffuse fibrosis in which there may be LV dilation.21
The anterior veil of the mitral valve is usually thickened and elongated. This mitral valve abnormality, along with the hypertrophy of the subaortic interventricular septum which protrudes towards the LV outflow tract, means that the anterior veil of the mitral valve contacts repeatedly in systole, producing a subaortic endocardial fibrous plaque, which is the mirror image of the anterior veil of the mitral valve. The mitral valve lesion causes reflux, with the increased size of the left atrium, which is the basis for the onset of atrial fibrillation.22
Microscopically, the myocytes are hypertrophic, with hyperchromatic nuclei, and are arranged in a disorganised manner, adopting a swirl or interwoven pattern. The architectural disorder affects the capillary network. The interstitium is highly increased, meaning that the myocytes are separated from each other, with a greater interstitial cellularity formed by fibroblasts visible. The intramyocardial arterial branches present their thickened and dysplastic walls (Fig. 2D), with fibrosis of the middle layer and irregular hyperplasia of the intimal cells which are laid out perpendicular to the vessel wall.21
The marked hypertrophy along with the architectural disorder and the intramural artery disease facilitate the onset of ischaemia and fibrosis. This fibrosis adopts two patterns: in the interstitial form the fibrosis is thin, around each myocyte; in the form called replacement fibrosis there are extensive areas with loss of myocytes which are replaced by collagen deposition. In the final phase of the disease, a reshaped ventricle with thinning of the ventricular wall and dilation of the cavity, similar to dilated cardiomyopathy, is produced if the fibrosis is very extensive.21 It is important to highlight that in normal hearts there is an area of architectural disorder in the anterior and posterior section of the interventricular septum in the ventricular-free wall junction.
The differential diagnosis with idiopathic left ventricular hypertrophy, which is a primary cardiac hypertrophy with no architectural disorder or fibrosis typical of HCM, should be carried out. It has been found in young people and athletes with no family history.12,17,21 Cardiologists consider it to be a variant of HCM.
Arrhythmogenic cardiomyopathyArrhythmogenic cardiomyopathy (ACM) is a genetic progressive disease of the myocardium characterised by replacement of the myocardium with fibroadipose tissue or fibrosis and which is clinically manifested by the onset of ventricular arrhythmias and SD in its first phase, and by heart failure in the advanced stage. It is a common cause of SD in individuals under the age of 35 years and in athletes in Spain.12,17 There are three forms: the right or classic form, the biventricular form with simultaneous compromise of the two ventricles and a form with predominant LV compromise which is more common in forensic pathology.23 The left form is a condition which is clinically under-recognised and is incorrectly diagnosed as dilated cardiomyopathy (DCM), HCM or viral myocarditis. Macroscopically, hearts are heavier and can have a globoid shape with dilatation of the RV or of both ventricles. In ACM of the RV, an aneurysm may form in the outflow tract. Aneurysms are not observed in the LV, probably due to the greater thickness of its wall. In cross sections of the ventricles, it is possible to see replacement of the myocardium with yellowish-greyish tissue in the subepicardial region of both ventricles, but which may be almost transmural in the RV, where only the trabeculae are preserved (Fig. 2E). This replacement may be focal, sometimes not very obvious to the naked eye, or be more extensive, affecting a large part of the free wall of both ventricles and the interventricular septum, where it adopts the form of a line deviating to the right. In the left-dominant forms, the posterolateral wall is preferentially affected, or there is circumferential compromise. A non-compaction pattern is sometimes adopted.21
The replacement of the myocardium by fibroadipose tissue is microscopically characteristic (Fig. 2F). In the left-dominant forms, the replacement may be almost exclusively fibrous, with limited adipose component. Some myocytes present apoptosis and cytoplasmic vacuolation, and it is common to find small inflammatory foci generally in preserved myocardium. In exceptional cases, the inflammation is extensive and subepicardial which, from a morphological point of view, cannot be differentiated from subacute myocarditis. The right form of ACM must be distinguished from lipomatosis, which does not present fibrosis.
Dilated cardiomyopathyDCM is characterised by increased ventricular volume, with reduced myocardial contraction force and systolic dysfunction. One third of cases of DCM die suddenly and it is more prevalent in males. The most common form of dilation of the heart in forensic pathology is that of ischaemic origin secondary to extensive transmural infarctions.
There are many aetiologies which cause DCM. Chronic consumption of alcohol and cocaine, infections and chemotherapy agents are the most common causes.24 It is estimated that 30% of DCMs are secondary to myocarditis.19 Between 30% and 40% of cases are genetic and may be familial.25 Alcoholic cardiomyopathy is an acquired form of DCM associated with the consumption of 80g of alcohol per day for more than five years,26 with greater susceptibility in women. Chronic alcoholism also results in cardiac hypertrophy, HTN and cardiac arrhythmias. In DCM, the heart has a globoid shape with increased weight (usually >500g in adults), dilated cavities, thinned walls, thickened endocardium and flattened trabeculae. There may be cavity thrombi which are generally located at the tip. The myocardium may look normal or present variable degrees of fibrosis, of irregular distribution (Fig. 2G).
The microscopic changes are highly varied and non-specific. There are cases in which the changes are minimal with thinned myocytes, with no increase in the interstitium. In others, the myocytes display great diversity in size, with atrophy and hypertrophy. The nuclei of the myocytes are hyperchromatic or multinucleated and there is interstitial and/or replacement fibrosis which varies in amount within the same case (Fig. 2H).
Left ventricular non-compaction cardiomyopathyThis cardiomyopathy is characterised by hypertrabeculation, meaning that the non-compacted area is double the size of the compacted area in the LV wall, compared to what occurs in a normal heart. It mainly affects the inferolateral wall of the middle third and apex of the LV. It may present in isolated form or associated with other cardiomyopathies, such as the cases that we have seen associated with ACM.21,27 It is a disease with heterogeneous genetics, with familial and sporadic forms, which mainly affects a wide age range of males.21
Heart valve diseaseIn our experience, it is responsible for 2% of SDs.6 Most cases correspond to aortic stenosis which may be located in the valve (the most common); above the sinotubular junction (supravalvular, associated with Williams syndrome) or subvalvular (membranous).28 Valve stenosis may be congenital (associated with the bicuspid aortic valve) or acquired. Bicuspid aortic valve is the most common congenital valve abnormality present in 1–2% of the population and mutations have been found in various genes that explain the familial aggregation.29
It is associated with coarctation of the aorta, interrupted aortic arch, dilated aorta, media degeneration and dissection, with aortic dissection being seven times more common than in the general population. Two asymmetric veils are identified. The bigger one has a central raphe or aborted commissure which does not reach the aorta wall and has elastic fibres, unlike the post-inflammatory fusion of a commissure. The raphe may be located in different positions but can most commonly be found between the right and left coronary sinuses, so that there is an anterior sinus, in which the origin of the two coronary arteries is located, and a posterior sinus. Early calcification of the valve from the age of 30 years (two decades before a normal valve) results in stenosis6,28 (Fig. 3A and B).
 Calcified bicuspid aortic valve. (B) Larger calcified raphe in left sinus which extends into the anterior veil of the mitral valve. (C) Mixed aortic stenosis: senile calcification with commissural fusion. (D) Chronic rheumatic mitral valve disease. (E) Infective endocarditis: vegetations on bioprosthesis (arrow). (F) Fibrin and platelets with numerous bacterial colonies (HE, 40×).")
(A) Calcified bicuspid aortic valve. (B) Larger calcified raphe in left sinus which extends into the anterior veil of the mitral valve. (C) Mixed aortic stenosis: senile calcification with commissural fusion. (D) Chronic rheumatic mitral valve disease. (E) Infective endocarditis: vegetations on bioprosthesis (arrow). (F) Fibrin and platelets with numerous bacterial colonies (HE, 40×).
Acquired aortic stenosis is mainly due to the degenerative calcification of the valve. It is the most common cause of valve replacement and of SD of valvular origin. It is a dynamic process of inflammation, accumulation of lipids and calcification similar to atherosclerosis, and the risk factors are similar. It is characterised by the presence of calcified masses at the base of the veils which limit the valve opening. Post-inflammatory stenosis tends to be of rheumatic origin, is much less common nowadays and is associated with mitral lesions. Over time it also calcifies, resulting in mixed forms (senile-post-inflammatory) (Fig. 3C), which, in very advanced cases, are indistinguishable from bicuspid valve stenosis.28
Regardless of the type of valvular lesion, aortic stenosis causes LV hypertrophy due to pressure overload, which leads to lesions of the intramyocardial arterioles responsible for angina symptoms and progressive fibrosis of half the thickness. LV hypertrophy tends to be concentric, but in 5% of cases it is asymmetric, similar to hypertrophic cardiomyopathy and, in the case of double valvular lesion, it may be eccentric.30
Mitral valve disease is much less significant in forensic practice. Lesions associated with age as well as the yellowish plaques in the anterior veil of the valve and ring calcification are the most common lesions.1 Rheumatic mitral valve disease is rare, as has already been mentioned, and we can see it in its chronic phase which is characterised by thickening of the veils, commissural fusion, shortening and thickening of the chordae tendineae and fibrosis of the papillary muscles28 (Fig. 3D).
Infective endocarditis may settle on normal valves or those with predisposing disease (rheumatic, myxomatous or calcific aortic stenosis). Bioprostheses are particularly susceptible, and mortality may reach 50% as a result of this complication. It is characterised by the presence of friable vegetations formed of fibrin, platelets and bacteria, on the free edge of the valves (Fig. 3E and F). The two main complications are necrosis and valve perforation, and embolisation of vegetation fragments which may become lodged in the coronary arteries, resulting in SD of ischaemic origin, or indirectly causing infarctions. Abscesses in the valve ring may spread to the myocardium. In patients with severe diseases, such as cancer, sepsis, etc., small vegetations (<5mm) may be seen along the valve closure line which are formed of fibrin with no bacteria or inflammatory cells; this is known as non-bacterial thrombotic endocarditis.28
Complications of prosthetic valves depend on the type of valve. In biological prostheses, in addition to the infective endocarditis discussed above, a tear in one of the veils, perforation and, over time, calcification, can occur. In mechanical prostheses, the most common complication is thrombosis and embolisation and valve movement dysfunction. Concentric, or pannus, overgrowth may occur in both types. However, the most common cause of SD in patients with prosthetic valves is associated cardiac hypertrophy which is accompanied by patches of fibrosis.28
Final considerationsThe specialised study of the heart makes it possible to explain with greater reliability SDs that occur in problematic circumstances (hospital environment or after a medical consultation) and to give advice to the family in cases with a genetic basis. This study may also have great social healthcare significance as it makes it possible to determine the most common aetiologies, the age and circumstances in which they occur and to take the appropriate preventive measures.
Conflicts of interestThe authors declare that they have no conflicts of interest.
Please cite this article as: Aguilera Tapia B, Suárez Mier MP. Diagnóstico post mortem de las cardiopatías estructurales. Rev Esp Med Legal. 2018;44:22–31.