










The phosphaturic mesenchymal tumour (PMT) is a very uncommon cause of oncogenic osteomalacia (OO), which is a paraneoplastic syndrome with severe clinical osteomalacia. The PMT is a neoplasia that produces the fibroblast growth factor FGF23, resulting in reduced proximal tubular phosphate reabsorption leading to hyperphosphaturia and hypophosphatemia. Our aim is to present our experience and complications in diagnosis and treatment of PMT in three patients.
Material and methodsWe propose an observational, descriptive and retrospective study of three cases of OO secondary to PMT found in our database of bone and soft tissue tumours. The inclusion criteria were: symptoms related with OO, presence of hyperphosphaturic hypophosphatemia, elevated levels of FGF23 in blood and pathological diagnosis of PMT.
ResultsIn all cases, the disease showed asthenia, non-specific bone pain, progressive functional weakness, and pathological fractures. The average delay time in diagnosis was 7 years. All presented with hyperphosphaturic hypophosphatemia, elevated levels of alkaline phosphatase as well as FGF23. The use of Octreoscan and PET-CT were essential to find the producing tumour and its subsequent biopsy. Treatment was surgery in two cases and one case was treated by CT-guided cryotherapy with neurophysiological control. Once the surgery was performed, the blood parameters normalized. There is no recurrence.
ConclusionsPhosphaturic mesenchymal tumor is a very rare entity as part of bone and soft tissue tumors, it may occur in both tissues. The phosphate-calcium homeostasis is altered due to high serum levels of FGF23 because of PMT. Delay in diagnosis is usual, leading to renal and skeletal comorbidities. To avoid this, knowledge of this entity together with high diagnostic suspicion are critical. Surgical treatment leads to normalization of serum levels and systemic symptoms.
El tumor mesenquimal fosfatúrico (TMF) es una causa poco frecuente de osteomalacia oncogénica (OO), síndrome paraneoplásico que puede cursar con clínica severa en el aparato locomotor. El TMF es una neoplasia mesenquimal productora del factor de crecimiento fibroblástico FGF23, a consecuencia del cual se produce hiperfosfaturia e hipofosfatemia resultando en el cuadro de OO. Nuestro objetivo es presentar nuestra experiencia y las complicaciones en el diagnóstico y tratamiento del TMF mostrando una serie de 3 casos.
Material y métodosPlanteamos un estudio observacional, descriptivo y retrospectivo de 3 casos de TMF como causa de OO encontrados tras búsqueda en el sistema de registro digital del hospital y nuestra base de datos de tumores del aparato locomotor. Los criterios de inclusión fueron los siguientes: cuadro clínico de OO, presencia de hipofosfatemia hiperfosfatúrica, niveles elevados de FGF23 en sangre y diagnóstico patológico de TMF.
ResultadosEn todos los casos, la enfermedad comenzo en forma de astenia, dolores óseos inespecíficos, limitación funcional progresiva y presencia de fracturas patológicas. El tiempo medio de retraso diagnóstico fue de 7 años. Analíticamente, todos presentaban una hipofosfatemia hiperfosfatúrica, elevación de los niveles de fosfatasa alcalina y de FGF23. La utilización del octreoscan y el PET/TAC fueron fundamentales para la localización del tumor productor y su posterior biopsia. El tratamiento fue la cirugía en 2 casos y un caso mediante crioterapia dirigida por TAC con control neurofisiológico. Una vez intervenidos se normalizaron los parámetros analíticos. No hemos registrado recidivas hasta la fecha.
ConclusionesEl TMF constituye una entidad rara en el campo de los tumores de partes blandas y óseos, ya que puede darse en ambos tipos de tejido. Debido a la producción del FGF23, ocasiona una alteración en la homeostasis fósforo-calcio. El retraso diagnóstico es la norma, lo cual conlleva a comorbilidades renales y esqueléticas. Para evitarlo, es preciso el conocimiento de la entidad junto con una alta sospecha diagnóstica. El tratamiento quirúrgico conlleva la normalización analítica y del cuadro sistémico.
The phosphaturic mesenchymal tumour (PMT) is a cause of oncogenic osteomalacia (OO), a paraneoplastic syndrome which may have severe clinical osteomalacia in the form of intense bone pains and multiple fractures.1 PMT produces high levels in the blood of FGF23,2 a hormone synthesised by this mesenchymal lesión.3,4 It is benign in general, with very varied anatomical location and may be located both in bones and soft tissues.5 Epidemiologically these lesions may appear at any age, with the peak of incidence the fifth and sixth decades of life.6 Diagnosis is generally late, with 5 and 6-year delays described in the literature, due to low prevalence, and subtle non-specific symptoms.7 Raised FGF23 levels lead to ongoing hypophosphataemia in relation to the renal-level phosphaturic action of this hormone.8 Diagnosis is essentially based on the detection of high levels of FGF23 in the blood,9 and demonstration with imaging tests such as octreoscan or PET/CT scan with 25-fluorodesoxyglucose.10 Treatment of the OO secondary to PMT, is based on the location and on the resection of the tumour responsible for causing the FGF23.11,12
Our objective was to present our experience and complications in the diagnosis and treatment of the PMT, from 3 cases treated in our centre, stressing the importance of metabolic phosphorous and calcium assessment and high suspected diagnosis.
Material and methodsThis was a retrospective study and analysis of progression in a series of three patients, identified after an exhaustive search in our orthopaedic oncology base, and in the tool Excalibur® from our hospital (it identifies any word written in discharge or surgical reports since 1978) using the terms “phosphaturic mesenchymal tumour ““oncogenic osteomalacia” and “hypophosphoraemia”. The inclusion criteria were the ones considered as diagnostic of this entity: clinical symptoms compatible with osteomalacia; analytical presence of hypophosphoraemia” (phosphorous >4,5 mg/dl) with hyperphosphaturia in urine of 24 h (>950 mg); raised levels of FGF23 in the blood (>3.5 U/I); determination of its existence using imaging tests in either conventional radiography (radiography, CT and NMR) or using nuclear medicine (octreoscan, bone scan and PET/CT with 25-fluorodesoxyglucose)and pathological diagnosis of the PMT (using hematoxylin-eosin tinctures). Epidemiological data (age, sex) were included, together with anamnesis (year of onset of symptoms, reason for referral to orthopaedic surgery), medical examination (presence of pathological fractures, location of them), radio diagnostic tests (radiography, CT or NMR), nuclear medicine tests (octreoscan or PET/CT with 25-fluorodesoxyglucose), laboratory tests with phosphorous, calcium, alkaline phosphatase (AF), parathyroid hormone (PTH), calcitriol and FGF23 levels.
Mean follow-up of patients was 52 months (range: 48–60 months).
ResultsWe now offer a description of the 2 cases (most relevant data are summarised in Tables 1–4).
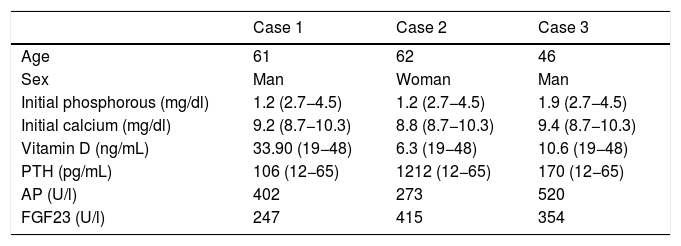
Epidemiological and laboratory data.
| Case 1 | Case 2 | Case 3 | |
|---|---|---|---|
| Age | 61 | 62 | 46 |
| Sex | Man | Woman | Man |
| Initial phosphorous (mg/dl) | 1.2 (2.7−4.5) | 1.2 (2.7−4.5) | 1.9 (2.7−4.5) |
| Initial calcium (mg/dl) | 9.2 (8.7−10.3) | 8.8 (8.7−10.3) | 9.4 (8.7−10.3) |
| Vitamin D (ng/mL) | 33.90 (19−48) | 6.3 (19−48) | 10.6 (19−48) |
| PTH (pg/mL) | 106 (12−65) | 1212 (12−65) | 170 (12−65) |
| AP (U/l) | 402 | 273 | 520 |
| FGF23 (U/l) | 247 | 415 | 354 |
AP: Alkaline Phosphatase; FGF23: Fibroblastic Growth Factor23; PTH: Parathyroid Hormone.
Clinical data of skeletal, systemic involvement and sequelae.
| Case 1 | Case 2 | Case 3 | |
|---|---|---|---|
| Location of pathological fractures | Right proximal femur | Vertebrae T11-T12-L1-L2 | Basicervical fractures of both femurs and vertebral compression |
| Systemic symptoms | General weakness | General weakness, asthenia | Progressive functional impotence |
| Sequelae | No | Chronic kidney failure and tertiary hyperparathyroidism | Bilateral arthroplasty of hip due to osteonecrosis |
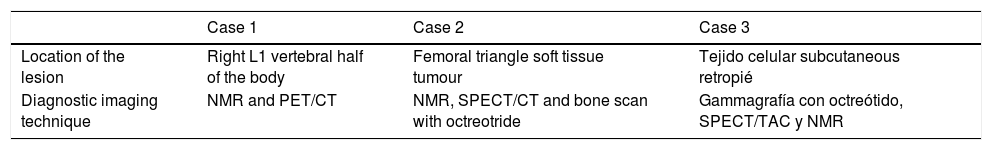
Imaging data.
| Case 1 | Case 2 | Case 3 | |
|---|---|---|---|
| Location of the lesion | Right L1 vertebral half of the body | Femoral triangle soft tissue tumour | Tejido celular subcutaneous retropié |
| Diagnostic imaging technique | NMR and PET/CT | NMR, SPECT/CT and bone scan with octreotride | Gammagrafía con octreótido, SPECT/TAC y NMR |
NMR: Nuclear Magnetic Resonance; PET/CT: Positron Emission Tomography/Computerised Axial Tomography; SPECT/CT: Single Photon Emission Computerised Tomography.
Sixty-one-year-old male, with no history of interest, assessed due to severe hypophosphataemia and treatment-resistant hyperphosphaturia. Clinically he suffered from general incapacitating bone pain and required crutches for walking. The images showed multiple bone lesions and fractures at several levels, with a prominent non-displaced sub capital fracture of the right femur. The PET/CT detected bilateral thyroid nodules with negative PAAF. The Tc-sestamibi marked bone scan presented focal uptake compatible with parathyroid adenoma, guiding diagnosis towards primary hyperparathyroidism (PHPT). A left hemithyroidectomy was performed resulting in normal parathyroid gland. Further analytic tests revealed hypophosphoraemia of 1.2 mg/dl, AF of 402 U/l and FGF23 of 247 U/l (n < 145). This finding guided diagnosis towards PMT, not initially detectable with NMR and PET/CT. A replacement medical treatment was selected, with the patient improving partially clinically and analytically. Due to the levels of FGF23 up to 357,2 U/l an octreoscan with no diagnosis was performed. Finally, repetition of the PET/CT detected an increase in metabolic activity in the right side of the body of L1 (Fig. 1). Following CT-guided needle core biopsy (NCB) a lesion compatible with PMT was diagnosed. Treatment consisted in percutaneous cryotherapy of the lesions guided by CT and simultaneous neurophysiologic control (Fig. 2). Phosphorous levels in blood and urine and FGF23 levels have normalised over time.


Sixty-two-year-old woman, with a history of left hemithyroidectomy due to nodular hyperplasia. Clinical symptoms began with general bone pain of 2 years onset, treated with NSAIDs. Following this, hypophosphataemia and hyperphosphaturia were found, AP of 273 U/l, severe radiographic osteopenia with vertebral T11-T12-L1-L2 fractures, with no trauma. Initially she was diagnosed with adult hypophosphoremic osteomalacia, and began medical treatment with phosphorous and calcitriol, which led to clinical improvement. After this, progressive impairment of renal function began with ongoing hypophosphataemia despite medical treatment. This led to chronic stage IV kidney disease with bilateral cortical nephrocalcinosis. Consecutively, elevation of PTH to 1.212 pg/mL, FGF23 to 415 U/l (n < 145), creatinine to 2.72 mg/dl and phosphorous to 1.7 mg/dl were detected. With these results, the octreoscan detected uptake in the right groin, with soft tissue mass at that level. After the NMR (Fig. 3) an ultrasound-guided CNB resulted in PMT. Extended resection was performed (Fig. 4) with histological confirmation of PMT, mad immediate analytical normalisation. Unfortunately this patient presented with chronic complications of hypophosphataemic rickets, SHPT and nephrocalcinosis.

.")
Forty-six-year old male, with no history of interest, with an onset of radiating low back pain in 2012 after effort. The initial NMR presented L5-S1 central vertebral disk hernia ion and L3-L4 left lateral and foramina vertebral disk herniation with no improvements of symptoms following conservative treatment. Decompression and instrumented arthodesis at L3-S1 levels was carried out in September 2013. His condition clinically worsened and he had difficulty walking, needing 2 crutches and finally a wheelchair. During follow-up we observed pseudo osteoarthritis at the levels where he had been operated on and an aseptic loosening of instrumentation. Further intervention took place in 2014, with removal of previous material, insertion of new material and the addition of a graft. Evolution was poor with a third operation required in June 2015. Intersomatic lumbar cages with a new autologous graft were added through anterior approach. Lack of improvement led to a bone metabolism study which showed hyperphosphaturic hypophosphataemia and raised FGF23 levels. A left femur neck fracture also occurred. The octreoscan revealed an increase in activity in the right heel footpad. The NMR confirmed soft tissue lesions (Fig. 5). Finally, given the small size of the lesion and high diagnostic suspicion, extended resection was performed, confirming PMT. The patient evolved favourably, with clinical and analyticla normalisation, and was able to perform daily life activities independently. Unfortunately he has required total bilateral hip replacements due to osteonecrosis at this level.

The main data from the 3 clinical cases presented are summarised in Tables 1–4.
DiscussionHypophosphataemia is a relatively common alteration, described in up to 5% of the total of hospitalised patients,13 the prolonged impairment of which gives rise to muscle changes and bone disorders. Thus when the clinical situation is of tumour origin, it may develop OO symptoms which, if severe and prolonged over time, may lead to major renal morbidity and morbidity of the locomotor system.14
In the 3 presented cases, the cause of OO was PMT, which may present at any age, and its appearance is more common between the fifth and sixth decade of life (Table 1). Diagnosis is generally late, with delays of 5 or 6 years being reported,6,7 which in our cases resulted in a mean delay of 80 months (Table 4). The first to report the existence of this entity was McCance15 in 1947, but it was Prader et al.16 in 1959, who recognised the tumour as the possible cause of osteomalacia. Most of these tumours usually settle in soft tissues or, to a lesser extent, in the bones of the lower limbs, trunk, head and neck. Microscopically2 they are composed of small, rounded, fussiform cells or spindle-shaped cells within a diffuse matrix that contains a delicate pattern of capillaries. The matrix tends to calcify, producing mineral deposits between the cells.
Diagnosis of the OO syndrome by PMT is simple but requires a high level of suspected diagnosis.17,18 We observed a severe osteomalacia, with significant hypophosphoraemia caused by unusually elevated phosphaturia (and therefore with a low rate of tubular reabsorption of phosphates [TRP]), calcium and normal PTH (at least initially), low level of calcitriol or inappropriately normal for hypophosphoraemia, significantly raised AP and resistance to treatment with vitamin D19 (Table 1). These facts are differential with regard to primary hyperpaththyroidism (PHPT), which generally involves hypercalcaemia, hypophosphataemia and raised serum levels of PTH. In contrast, SHPT due to vitamin D deficit has normal levels of calcium. Also, in SHPT to chronic kidney failure we find hypocalcaemia, hyperphosphoraemia, elevated levels of PTH and low levels of calcitriol.20 After this, the syndrome diagnosis was confirmed with a very high serum level of FGF239 (Table 1). When the symptoms appear in early ages, differential diagnosis with hypophosphoremic rickets should be made, and for this the presence or non presence of family members with a similar syndrome is of relevance, together with the presence or non presence of a mesenchymal tumour.21 High levels of FGF23 with hypophosphoraemia and hypophosphaturia are also produced in McCune-Albright22 syndrome and in epidermic nevus syndrome,23 but in both cases the cutaneous lesions and other manifestations are evident from the onset. Furthermore, relapses of the disease seem to be related to levels of FGF23 in the blood and phosphorous in urine.24
In our patients (Table 1), we observed a major hypophosphataemia with serum levels around 1.2–1.9 mg/dl, together with osteomalacia symptoms characterised systemically by major asthenia and skeletal clinical symptoms consistent with pathological fractures or femoral avascular necrosis (Table 2), which led to functional impairment for walking. Separately, chronic complications derived from hypophosphataemic rickets, such as hyperparathyroidism and nephrocalcinosis19 appeared in one of our cases (Table 2).
Characteristically, as confirmed in our cases, this osteomalacia is resistant to vitamin D treatment and whilst it is not detected and hypophosphataemia increases, the symptoms are not corrected.6 In the patients in our series, it took a long time to specify the medical treatment of the hypophosphataemia. When established a slow, but steady recovery from osteomalacia and its concomitant morbidity was obtained, but with ongoing hyperphosphaturia, the persistence of the medical symptoms desisted. Even so, symptomatic treatment does not diagnose or treat the cause of the problem, which requires identifying high serum levels of FGF23 and the anatomical location of the tumour producing them.25 The determination of FGF23 using ELISA techniques has been calculated to enable clinical diagnosis confirmation with an estimated sensitivity of 23%–86% with the tumour unidentified and of 38%–100% with the tumour confirmed.24 However, cases of PMT with normal FGF23 have been described.26 In all our cases, once suspicion of PMT was present, within the differential diagnosis of hypophosphatemic rickets, very high levels of FGF23 (Table 1) were observed.
The anatomical location of the tumour requires a detailed physical examination, together with imaging tests: octreoscan, PET/CT with 25-fluorodesoxyglucose and NMR.10 Also, in recent years new imaging tests have been included, such as Ga-DOTANOC PET/CT (using a modified octeotride molecule) or venous sampling with FGF23 in areas where imaging studies suggest suspicious lesions.25,10 In our centre we chose to use the octreoscan and PET/CT with 25-fluorodesoxyglucose for anatomical location of the tumour and then the NMR to identify and schedule biopsy and treatment. Even so, due to the size with which on occasions the PMT presents, it is not possible to initially detect it by these methods, and repetition of imaging tests is therefore recommended during follow-up,27 which also occured in our cases (Table 3).
Definitive PMT treatment consists in surgical resection,28 which is followed by a fast analytical improvement and the consequent regression of symptoms. Cases 2 and 3 were surgically treated with extended resection. Case 1 opted for percutanoue ablation with CT-guided cryotherapy under neurophysiologic control. This technique is widely used for the percutaneous treatment of benign mesenchymal tumours, including PMT.29–32 Based on the experience of our group, we believe that this therapeutic model has major advantages in both the eradication of the tumour and also in minimising complications and aiding patients’ recovery when anatomical locations are complex.
Several cases of PMT have been described in the literature, with histological signs of malignancy and the development of metastasis, but this is very uncommon33–35 and has not been our experience. Due to this, and to the possible reappearance of symptoms from local tumour relapse, the follow-up of these patients has to be long-term. Furthermore, after treatment, the function prognosis is generally good, except in those severe renal complications or skeletal complications where regression is not possible.27 Our patients completely recovered from the osteomalacia, with the sequelae of the same having to be treated, as well as pathological fractures and hip osteonecrosis (Table 2). After surgery, all our patients experienced normalising of FGF23 and phosphorus levels in blood and urine. As Jonsson et al. described in their article in 2003, postoperative increases in FGF23 levels is usually interpreted as local level tumour recurrence.26 In our case no relapse has to date presented, although follow-up is short (Table 4). In those rare cases in which the tumour producing FGF23 is not identified, treatment is based on replacement therapy with phosphorus and calcitriol supplements,36 or possible usage of burosumab. This drug, a monoclonal anti-FGF23 antibody, has only been currently approved by the FDA for regular use in the treatment of hypophosphataemia linked to chromosome X,37 although specific clinical trials were already in existence in the indication of FGF23 in managing oncogenic osteomalacia.38,39
Our study has major limitations. It is a retrospective study with casuistic of 3 cases and a follow-up which has been limited to date. Most of the references consulted present very limited casuistics,25,40–42 with the most numerous series to date being that published by Fang et al. in 2017 with 144 Cases,27 although collected over an interval of 32 years, which resulted in a mean of approximately 4 cases per year. Regarding study design, we would stress that some of the most impacting studies reviewed,2,11,27 are retrospective in design which are at an advantage in the context of rare pathologies. Regarding follow-up times, with the exception of the 2017 study by Feng et al. 27 that has higher follow-up times, there are published articles with similar follow-up times to ours.11 To sum up, our observations are similar to those of the previous different publications, regarding delay in diagnosis, clinical and analytical presentation, pathological anatomy and diagnostic methods, treatment, evolution and follow-up. Therefore, due to the rarity of this entity, we believe its communication is important given the clinical implications of morbidity caused by PMT since unfortunately delayed diagnosis is commonplace.
ConclusionPMT is an extremely rare nosologic entity, which causes hypophosphataemic and hyperphosphaturic OO syndrome resistant to treatment with vitamin D. Its rarity as an entity and its subtle, non-specific symptoms enable it to go unnoticed among the different specialities which cover this pathology: orthopaedic surgery, endocrinology, nephrology, radiodiagnostics and rheumatology. This entity must therefore be included in differential diagnosis when hypophosphataemic rickets are present, with the performing of an exhaustive phosphorous and calcium metabolic study, obtaining FGF23levels and carrying out imaging studies (octreoscan, PET/TC and NMR), to rule out or identify the presence of PMT. PMT resectioning is the first therapeutic option. When the tumour is located in complex anatomical situations for surgery or the morbidity it could lead to, percutaneous treatment with radiofrequency or CT-guided cryotherpay is a plausible option. On suspicion of this entity or its confirmation, we would recommend referral to centres of reference.
Level of evidenceLevel of evidence IV.
Conflict of interestsThe authors have no conflict of interests to declare.
To Dr. D. Ignacio Sánchez del Campo, a national level pioneer of tumoral musculoskeletal diseases.
Please cite this article as: Moreno Romero M, Pérez Muñoz I, González Lizán F, Gallego Rivera JI, Valdivielso Cañas L. El tumor mesenquimal fosfatúrico como causa de osteomalacia oncogénica. A propósito de 3 casos y revisión de la literatura. Rev Esp Cir Ortop Traumatol. 2021;65:442–449.
