



Several studies have reported increased oxidation of lipids, proteins and DNA in the brains of patients with Alzheimer disease (AD). Moreover, these patients display differences in the activity and polymorphisms of the genes encoding the enzymes GST (T1, M1) and MnSOD. For these reasons, we designed a study of the variability in GSTT1, GSTM1, and MnSOD genes in healthy and AD groups from a Venezuelan population.
MethodsWe included 179 unrelated Venezuelan subjects classified as either AD patients (n=79) or healthy individuals (n=100). Presence or absence of the GSTT1/GSTM1 genes was determined using PCR-SSP, and polymorphisms of MnSOD and APOE genes were identified with PCR-RFLP.
ResultsThe genotype GSTT1+/GSTM1− seems to favour development of AD (OR=2.06, P=.01). The risk level is higher when it is combined with the ¿4 allele of the APOE gene: GSTT1+/GSTM1−/¿3¿4 (OR=3.07, P=.05), GSTT1+/GSTM1−/¿4¿4 (OR=5.52, P=.02). The Ala-9Val polymorphism does not appear to be related to AD. However, the presence of the Ala/Ala genotype increases the risk provided by the ¿4 allele of the APOE gene: AlaAla/¿3¿4 (OR=3.47, P=.03), AlaAla/¿4¿4 (OR=6.3, P=.01).
ConclusionsThe results support the hypothesis that impaired mitochondrial function and increased oxidative damage are involved in the pathogenesis of AD. It is important to study other genes related to oxidative stress and antioxidant pathways which could be involved in susceptibility to AD.
Diversos estudios han descrito que en los cerebros de pacientes con enfermedad de Alzheimer (EA) hay una mayor oxidación de lípidos, proteínas y ADN. Además, en estos pacientes se ha observado diferencias en la actividad y polimorfismos de los genes que codifican las enzimas GST (T1 y M1) y MnSOD. En virtud de ello se planteó estudiar la variabilidad de los genes GSTT1, GSTM1 y MnSOD en individuos venezolanos sanos y con EA.
MétodosSe incluyeron 179 individuos venezolanos, no relacionados, agrupados en pacientes con EA (n=79) e individuos sanos (n=100). La presencia o ausencia de los genes GSTT1/GSTM1 se determinó por PCR-SSP y los polimorfismo de los genes MnSOD y APOE por PCR-RFLP.
ResultadosEl genotipo GSTT1+/GSTM1− parece favorecer el desarrollo de la EA (OR=2,06; p=0,01), siendo el riesgo mayor al estar en combinación con el alelo ¿4 del gen APOE: GSTT1+/GSTM1−/¿3¿4 (OR=3,07; p=0,05), GSTT1+/GSTM1−/¿4¿4 (OR=5,52; p=0,02). El polimorfismo Ala-9Val por sí solo no parece estar relacionado con la EA, sin embargo, la presencia del genotipo Ala/Ala incrementa el riesgo que proporciona el alelo ¿4 del gen APOE: AlaAla/¿3¿4 (OR=3,47; p=0,03), AlaAla/¿4¿4 (OR=6,3; p=0,01).
ConclusionesLos resultados apoyan la hipótesis de que el deterioro de la función mitocondrial y el aumento de daño oxidativo están involucrados en la patogénesis de la EA. Es importante estudiar otros genes relacionados con estrés oxidativo y vías antioxidantes, los cuales pudiesen estar involucrados en la susceptibilidad a desarrollar la EA.
Neurodegenerative diseases, including Alzheimer disease (AD), display oxidative damage as a common feature, but it is unclear whether this process is the cause or the effect of the disease.1 Oxidative damage may be caused by the peptide amyloid β (Aβ) and the paired helical filaments of tau protein that constitute the typical lesions in AD. The Aβ peptide has been observed to give rise to free radicals2 and inhibit the cytochrome oxidase enzyme. This contributes to oxidative stress.3 Observations have also shown that the paired helical filaments of tau protein may form advanced glycation end-products, and this generates enough reactive oxygen species (ROS) to cause oxidative damage.4 Since the brain is a major consumer of oxygen with a high demand for energy and only limited antioxidant ability compared to other tissues, it is very susceptible to oxidative damage. Antioxidant defences therefore play a vital role as the means of eliminating free radicals.5 Patients with AD have displayed altered activity of glutathione S-transferase enzymes (GSTs) and manganese superoxide dismutase (MnSOD).6 GSTs are a family of enzymes that are crucial to the protection of cells against toxic substances and oxidative stress. They are encoded by some 16 genes, further subdivided into 8 classes.7 The μ class comprises 5 different isoenzymes named GSTM1 through GSTM5. The θ class includes only 2 isoenzymes, GSTT1 and GSTT2. The null genotypes of genes GSTM1 and GSTT1 are characterised by homozygous knockout of the gene, resulting in absent enzyme activity (overview by Cooper8). Both the GSTT1 and GSTM1 enzymes are known for their ability to catalyse the detoxification of reactive oxygen and products of lipid peroxidation.9 Because of this, inactivity of the GSTT1/M1 enzymes has been linked to greater exposure to oxidative stress.10
The superoxide dismutase enzyme (SOD) is one of the main defences against the damage that can be caused by the free radical superoxide (O2−). The enzyme catalyses the conversion of superoxide into molecular oxygen (O2) and hydrogen peroxide (H2O2); these molecules will later be converted into water by the action of catalase or glutathione peroxidase enzymes.11 Superoxide dismutase exists in 3 isoforms: cytosolic [Cu/ZnSOD], extracellular [EC-SOD], and mitochondrial [MnSOD].12 Since 90% of all ROS originate in the mitochondria, MnSOD is an antioxidant which plays a fundamental role in protecting cells against oxidative stress. The activity of this mitochondrial enzyme defends cells from lipid peroxidation; within the brain, it protects the viability of the neuronal membrane.13 MnSOD enzyme is synthesised in the cytosol and then transported to the mitochondria. MnSOD transport requires the participation of a sequence of 24 amino acids known as the mitochondrial targeting sequence (MTS). These amino acids form the amphiphilic helices required to transport the enzyme to the mitochondria.14 The gene that encodes MnSOD possesses a polymorphism by which thymine (T) is replaced by cytosine (C) at nucleotide 47, resulting in an amino acid substitution (Val→Ala) at position −9 of the MTS and altering the enzyme structure. These changes in the structure alter mitochondrial transport of MnSOD, and therefore affect the cell's ability to defend itself from superoxide.14 Multiple studies have now demonstrated the role MnSOD plays in how neurons survive oxidative stress.15,16 Considering that GSTT1, GSTM1, and MnSOD enzymes contribute to cell defence against oxidative stress, and the possibility that oxidative stress and AD pathogenesis are related, we aim to study the role of polymorphisms of the genes GSTT1, GSTM1 and MnSOD in the development of AD and correlate any such associations with the presence of allele ¿4 of the APOE gene.
Materials and methodsPatientsThe study was carried out in 79 patients (mean age, 70±10 years), all of whom had been diagnosed with sporadic AD. All patients had received treatment in the neurology department at Hospital Clínico Universitario de Caracas (Venezuela) at some time between September 2004 and October 2006. These patients were selected according to the clinical protocol implemented in the Luis Borges Neuropsychology Unit of the neurology department at Hospital Clínico Universitario de Caracas. The protocol was framed in accordance with the specifications of the American Psychiatric Association (DSM-IV) and the NINCDS-ADRDA Alzheimer's Criteria (National Institute of Neurological Disorders, Communicative Disorders, and Stroke; Alzheimer's Disease and Related Disorders Association).
The control group consisted of 100 healthy Venezuelan residents with a mean age of 71±10 years. These subjects completed the Mini-Mental State Examination and laboratory and imaging studies.
All participants in the study gave their informed consent; the consent of participants with AD was authorised by their legal guardians. The consent procedure was approved by the bioethics committees at Instituto Venezolano de Investigaciones Científicas and Hospital Clínico Universitario de Caracas.
Genomic DNA extractionGenomic DNA was extracted from leukocytes and lymphocytes in peripheral blood according to Bunce's method.17
Detection of GSTT1/GSTM1 genesA multiplex PCR protocol was used to determine presence or absence of the GSTM1 and GSTT1 genes according to a modified version of the method described by Lin et al.18 and the specific initiators used by Chen et al.19
Genotyping of the MnSOD geneThe Ala-9Val polymorphism was studied using the PCR-RFLP method with the initiators and protocol described by Ambrosone et al.20
Genotyping of the APOE geneResearchers genotyped and evaluated the APOE polymorphism using PCR-RFLP with the initiators described by Emi et al.21 and the protocol published by Hixson and Vernier.22
Statistical analysisGenotypic and allelic frequencies were calculated. The statistical significance of intergroup differences in frequency (for alleles, genotypes, and genotypic combinations) was estimated using the Mantel–Haenszel chi-square statistic and 2×2 contingency tables. Researchers adjusted P-values by multiplying them by the number of completed comparisons (Bonferroni correction). Differences were considered statistically significant for P<.05.
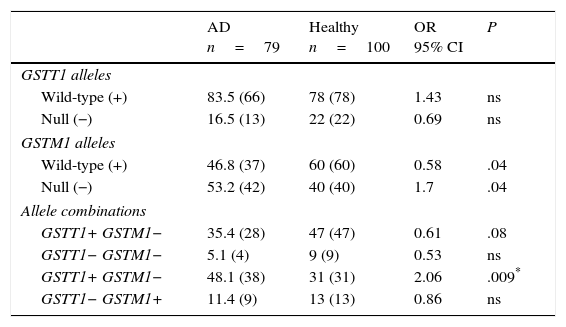
ResultsGenotype distribution and genotypic combination of GSTT1 and GSTM1 genes in healthy individuals and patients with Alzheimer diseaseTable 1 displays the distribution of the GSTM1 (wild-type [+], null [−]) and GSTT1 (wild-type [+], null [−]) in healthy individuals and patients with AD. The comparison of frequencies revealed the wild-type GSTM1 genotype to be significantly more frequent in controls than in patients (OR: 0.58; 95% CI: 0.3235-1.0659; P=.04; Pcorr=ns). In contrast, the null GSTM1 genotype had a significantly higher frequency in patients than in controls (OR: 1.7; 95% CI: 0.9381-3.0903; P=.04; Pcorr=ns). Nevertheless, differences were not statistically significant once P-values were corrected.
Distribution of genotypes and genotype combinations for the genes GSTT1 and GSTM1 in patients with AD and healthy controls.
| AD n=79 | Healthy n=100 | OR 95% CI | P | |
|---|---|---|---|---|
| GSTT1 alleles | ||||
| Wild-type (+) | 83.5 (66) | 78 (78) | 1.43 | ns |
| Null (−) | 16.5 (13) | 22 (22) | 0.69 | ns |
| GSTM1 alleles | ||||
| Wild-type (+) | 46.8 (37) | 60 (60) | 0.58 | .04 |
| Null (−) | 53.2 (42) | 40 (40) | 1.7 | .04 |
| Allele combinations | ||||
| GSTT1+ GSTM1− | 35.4 (28) | 47 (47) | 0.61 | .08 |
| GSTT1− GSTM1− | 5.1 (4) | 9 (9) | 0.53 | ns |
| GSTT1+ GSTM1− | 48.1 (38) | 31 (31) | 2.06 | .009* |
| GSTT1− GSTM1+ | 11.4 (9) | 13 (13) | 0.86 | ns |
AD: Alzheimer disease; 95% CI: confidence interval; ns: not significant; OR: odds ratio.
Values shown in parentheses indicate the number of individuals bearing the genotype for the polymorphic site under study. Frequencies are expressed as percentages.
* Significant after applying the Bonferroni correction.
The study of genotype combinations for GSTT1 and GSTM1 found that all possible combinations were present in both groups. The GSTT1+/GSTM1− combination was the most frequent in patients with AD, followed by the combinations GSTT1+/GSTM1+, GSTT1−/GSTM1+, and GSTT1−/GSTM1−. The GSTT1+/GSTM1+ combination was the most frequent among healthy controls, followed by the combinations GSTT1+/GSTM1−, GSTT1−/GSTM1+, and GSTT1−/GSTM1−. The comparison of frequencies revealed the combination GSTT1+/GSTM1− to be significantly more frequent in patients than in controls (OR: 2.06; 95% CI: 1.1118-3.8036; P=.09; Pcorr=.036). The P-value remained significant after correction (Table 1).
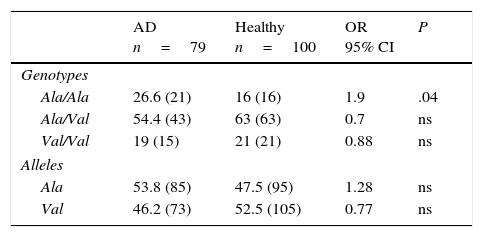
Allelic and genotypic frequencies of the MnSOD gene in healthy individuals and patients with Alzheimer diseaseIn both groups, the Ala/Val genotype had the highest frequency, followed by Ala/Ala and Val/Val in AD patients and Val/Val and Ala/Ala in healthy controls. The comparison of frequencies revealed the Ala/Val genotype to be significantly more frequent in patients than in controls (OR: 1.9; 95% CI: 0.9147-3.9498; P=.04; Pcorr=ns). However, differences were not statistically significant once P-values were corrected (Table 2).
Allelic and genotypic frequency of the Ala-9Val polymorphism of the MnSOD gene in patients and controls.
| AD n=79 | Healthy n=100 | OR 95% CI | P | |
|---|---|---|---|---|
| Genotypes | ||||
| Ala/Ala | 26.6 (21) | 16 (16) | 1.9 | .04 |
| Ala/Val | 54.4 (43) | 63 (63) | 0.7 | ns |
| Val/Val | 19 (15) | 21 (21) | 0.88 | ns |
| Alleles | ||||
| Ala | 53.8 (85) | 47.5 (95) | 1.28 | ns |
| Val | 46.2 (73) | 52.5 (105) | 0.77 | ns |
AD: Alzheimer disease; 95% CI: confidence interval; ns: not significant; OR: odds ratio.
Values shown in parentheses indicate the number of repeats of the allele or the number of individuals bearing the genotype for the polymorphic site under study. Frequencies are expressed as percentages.
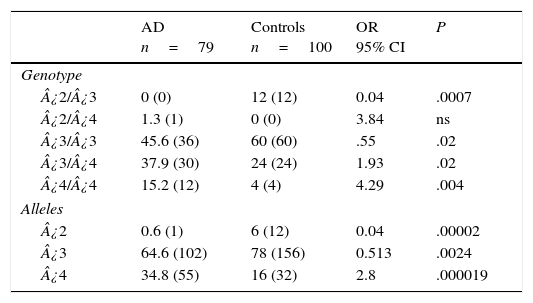
Table 3 displays the genotypes of the APOE gene for AD patients and healthy controls. The frequencies of genotypes ¿2¿3 (OR=0.04; 95% CI: 0.002-0.764; P=.0007; Pcorr=.0035) and ¿3¿3 (OR=0.55; IC 95%: 0.3073-1.0136; P=.02; Pcorr=ns) were significantly higher in controls than in AD patients. In contrast, patients displayed higher frequencies of genotypes ¿3¿4 (OR=1.93; 95% CI: 1.0164-3.6980; P=.02; Pcorr=ns) and ¿4¿4 (OR=4.29; 95% CI: 1.3241-13.9016; P=.004; Pcorr=.02). The comparison of allele frequencies also showed that ¿2 (OR=0.04; 95% CI: 0.0059-0.3651; P=.000025; Pcorr=.000075) and allele ¿3 (OR=0.5; 95% CI: 0.3220-0.8195; P=.0024; Pcorr=.0072) were significantly more common among controls than in AD patients. Nevertheless, allele ¿4 was significantly more frequent in AD patients than in controls (OR=2.8; 95% CI: 1.7003-4.6221; P=.000019; Pcorr=.000057).
Frequencies of genotypes and alleles of the APOE gene in patients with AD and controls.
| AD n=79 | Controls n=100 | OR 95% CI | P | |
|---|---|---|---|---|
| Genotype | ||||
| ¿2/¿3 | 0 (0) | 12 (12) | 0.04 | .0007 |
| ¿2/¿4 | 1.3 (1) | 0 (0) | 3.84 | ns |
| ¿3/¿3 | 45.6 (36) | 60 (60) | .55 | .02 |
| ¿3/¿4 | 37.9 (30) | 24 (24) | 1.93 | .02 |
| ¿4/¿4 | 15.2 (12) | 4 (4) | 4.29 | .004 |
| Alleles | ||||
| ¿2 | 0.6 (1) | 6 (12) | 0.04 | .00002 |
| ¿3 | 64.6 (102) | 78 (156) | 0.513 | .0024 |
| ¿4 | 34.8 (55) | 16 (32) | 2.8 | .000019 |
AD: Alzheimer disease; 95% CI: confidence interval; ns: not significant; OR: odds ratio.
Values in parentheses indicate the number of individuals bearing the genotype. Frequencies are expressed as percentages.
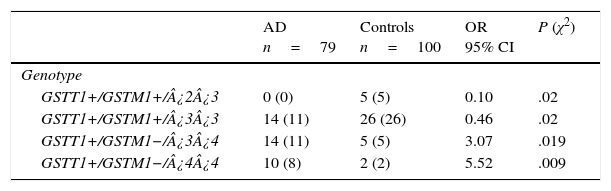
There are 24 possible combinations of GSTT1/GSTM1/APOE, and we observed 11 in the AD group and 15 in the controls. The frequencies of the combinations GSTT1+/GSTM1+/¿2¿3 (OR=0.1; 95% CI: 0.0059-2.004; P=.02; Pcorr=ns) and GSTT1+/GSTM1+/¿3¿3 (OR=0.46; 95% CI: 0.2114-1.002; P=.02; Pcorr=ns) were significantly higher in controls than in patients. In contrast, frequencies of the combinations GSTT1+/GSTM1−/¿3¿4 (OR=3.07; 95% CI: 1.0210-9.2514; P=.019; Pcorr=ns) and GSTT1+/GSTM1−/¿4¿4 (OR=5.52; 95% CI: 1.1381-26.7837; P=.009; Pcorr=ns) were significantly higher in AD patients than in controls (Table 4).
Frequencies of genotype combinations for GSTT1/GSTM1/APOE in patients with AD and controls.
| AD n=79 | Controls n=100 | OR 95% CI | P (χ2) | |
|---|---|---|---|---|
| Genotype | ||||
| GSTT1+/GSTM1+/¿2¿3 | 0 (0) | 5 (5) | 0.10 | .02 |
| GSTT1+/GSTM1+/¿3¿3 | 14 (11) | 26 (26) | 0.46 | .02 |
| GSTT1+/GSTM1−/¿3¿4 | 14 (11) | 5 (5) | 3.07 | .019 |
| GSTT1+/GSTM1−/¿4¿4 | 10 (8) | 2 (2) | 5.52 | .009 |
AD: Alzheimer disease; 95% CI: confidence interval; OR: odds ratio; χ2: chi-square test.
Values in parentheses indicate the number of individuals bearing the genotype. Frequencies are expressed as percentages.
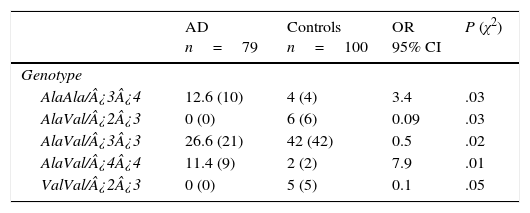
Of the 18 possible combinations of MnSOD/APOE, 12 were observed in controls and 9 in patients. The combinations AlaAla/¿3¿4 (OR=3.4; 95% CI: 1.0476-11.5486; P=.016; Pcorr=ns) and AlaVal/¿4¿4 (OR=7.9; 95% CI: 1.7027-36.8988; P=.001; Pcorr=ns) were significantly more common in AD patients than in controls. We must point out that some genotype combinations were present only in the control group: AlaVal/¿2¿3 (OR=0.09; 95% CI: 0.0050-1.6480; P=.014; Pcorr=ns), AlaVal/¿3¿3 (OR=0.5; 95% CI: 0.2642-0.9461; P=.016; Pcorr=ns), and ValVal/¿2¿3 (OR=0.1; 95% CI: 0.0059-2.0049; P=.02; Pcorr=ns) (Table 5).
Frequencies of genotype combinations of the MnSOD/APOE gene in patients with AD and controls.
| AD n=79 | Controls n=100 | OR 95% CI | P (χ2) | |
|---|---|---|---|---|
| Genotype | ||||
| AlaAla/¿3¿4 | 12.6 (10) | 4 (4) | 3.4 | .03 |
| AlaVal/¿2¿3 | 0 (0) | 6 (6) | 0.09 | .03 |
| AlaVal/¿3¿3 | 26.6 (21) | 42 (42) | 0.5 | .02 |
| AlaVal/¿4¿4 | 11.4 (9) | 2 (2) | 7.9 | .01 |
| ValVal/¿2¿3 | 0 (0) | 5 (5) | 0.1 | .05 |
AD: Alzheimer disease; 95% CI: confidence interval; OR: odds ratio; χ2: chi-square test.
Values in parentheses show the number of individuals bearing the genotype for the polymorphic site under study. Frequencies are expressed as percentages.
The imbalance between the production and the elimination of ROS and nitrogen is known as oxidative stress. The cell must overcome oxidative stress to restore redox balance and thereby avoid the neuronal death and loss of neuronal function that are associated with neurodegenerative diseases.23 Studies in the area of AD pathogenesis have focused on the role of oxidative stress since the central nervous system is a great consumer of energy with only minimal antioxidant defences, making it highly sensitive to oxidative stress.24 In the brain with AD, antioxidant systems function less effectively, which may lead to an increase in the reactive species of oxygen and nitrogen that would react to biomolecules, including proteins, lipids, carbohydrates, DNA, and RNA. This would cause changes in their chemical structure, and in turn, loss of function.25 Oxidative stress in the brains of AD patients is well-documented: researchers have observed high levels of antioxidant enzymes, as well as increased concentrations of oxidative stress markers in the brains of AD patients compared to those of age-matched controls. Furthermore, the role played by oxidative stress in AD progression was observed when we compared the presence of the same brain proteins with oxidative damage in subjects with mild cognitive impairment, early-stage AD, and late-stage AD. This demonstrates that certain major pathways are activated and may be involved in AD progression.25 Given that the GST family of enzymes conjugates reduced glutathione with electrophilic compounds, thus helping remove it from cells to prevent oxidative damage,26 several studies have been published on the genes that encode the GSTM1 and GSTT1 enzymes and their role in different diseases. These enzymes proceed from a null phenotype characterised by absence of enzymatic activity due to inherited homozygous elimination of the full gene. In this study, the null GSTM1 genotype was significantly more frequent in patients with AD (OR: 1.7; P=.04; Pcorr=ns); the wild-type GSTM1 genotype was significantly more common in healthy controls (OR=0.58; P=.04; Pcorr=ns), in contrast with findings from other studies.9,27–30 Likewise, the combination GSTT1+/GSTM1− was more frequently seen in AD patients than in controls (OR: 2.06; Pcorr=.036). These results suggest that the null genotype GSTM1 and the combination GSTT1+/GSTM1− could confer up to twice the risk of developing AD. Considering that a variety of studies suggest that variations in the APOE gene may be genetic risk factors for AD,31–33 we recorded the combinations of GSTT1/GSTM1 and APOE genotypes and obtained interesting results. The genotype combinations GSTT1+/GSTM1−/¿3¿4 and GSTT1+/GSTM1−/¿4¿4 were significantly more frequent in patients than in controls. These combinations may therefore be linked to increased likelihood of developing AD. Additionally, the risk posed by the presence of one or 2 ¿4 alleles of the APOE gene is higher in the combination in which the GSTM1 gene is absent. Considering the above, an increase in Aβ peptide aggregation due to the E4 isoform of apolipoprotein E,34 together with the reduction in antioxidant defences caused by homozygotic elimination of the GSTM1 gene, could generate greater oxidative stress. This in turn would give rise to more neuronal death and explain how the risk inherent to allele ¿4 of the APOE gene would multiply in the absence of the GSTM1 gene. Furthermore, combinations including wild-type GSTT1 and GSTM1 and at least one ¿3 allele [GSTT1+/GSTM1+/¿2¿3 and GSTT1+/GSTM1+/¿3¿3] were significantly more frequent in healthy controls than in AD patients. This suggests that these combinations may provide protection against the onset of AD. Aβ peptides are components of the senile plaques that begin degeneration of brain neurons in AD by increasing the presence of ROS until it exceeds the defence ability of each cell. Kaminsky and Kosenko35 studied the effects of Aβ peptides on antioxidant enzymes from mitochondrial and non-mitochondrial sources in the rat brain in vivo. They observed that Aβ peptides increase the enzymatic activities creating H2O2 while inhibiting the activity of enzymes that consume H2O2 in the mitochondria and cytosol. This finding suggests that the imbalance between enzymes generating and those metabolising H2O2 contribute to the underlying oxidative stress in the neurodegeneration and neuronal death that occur in AD. Furthermore, the Aβ peptide has been found to be involved in the decreased expression of cytochrome c oxidase in the mitochondria, which in turn affects the electron transport chain and produces ROS.3 The MnSOD enzyme forms part of the cell's enzymatic antioxidant defences, and in fact provides the cell's first line of defence against the superoxide anion by converting it into H2O2.12 Multiple studies now support that MnSOD plays a role in how neurons survive oxidative stress. One such study, performed in MnSOD gene-knockout mice, reported that the mice died not long after birth with signs of neurodegeneration.15,16 Similarly, studies in transgenic rats have reported that MnSOD deficiency increases the concentrations of the Aβ peptide, which favours the formation of senile plaques.36 However, other studies have reported that overexpression of the MnSOD enzyme actually protects neurons from oxidative damage.37–39 In light of this conflicting evidence, we studied the Ala-9Val polymorphism of the MnSOD gene that alters the secondary structure of that protein, and therefore the mitochondrial transport of MnSOD. This affects the cell's ability to defend itself from superoxide radicals.40 A comparison of the frequencies of the Ala-9Val genotypes of MnSOD showed that genotype Ala/Ala was significantly more frequent in patients than in healthy controls. This finding does not coincide with the results from an Italian population reported by Ventriglia et al.13 Furthermore, evaluating the combined effect of the MnSOD and APOE genes revealed that the frequencies of the combinations AlaAla/¿3¿4 (OR=3.4; P=.03) and AlaVal/¿4¿4 (OR=7.9; P=.01) were significantly higher in patients than in controls, meaning that these combinations may indicate a greater susceptibility to AD. The presence of the Ala allele, which is associated with greater enzymatic activity of MnSOD in humans, as well as the presence of allele ¿4, promotes the development of AD. We should point out that the risk associated with genotypes ¿3¿4 and ¿4¿4 (OR=1.93 and OR=4.29, respectively) is approximately twice as high when the genotypes AlaAla and AlaVal of the MnSOD gene are present (OR=3.4 and OR=7.9, respectively). In contrast, the genotypic combinations AlaVal/¿2¿3, AlaVal/¿3¿3, and ValVal/¿2¿3 were only present in healthy individuals. This suggests that they may play a protective role against AD. Allele ¿3, whether present homozygously or with allele ¿2, together with one or 2 doses of the Val allele that is associated with abnormal enzymatic activity by MnSOD,40,41 would therefore protect individuals from developing AD. Considering that the Ala form of MnSOD is more efficiently transported to the mitochondria than the Val form,14 and that this enzyme catalyses the dismutation of the superoxide anion in H2O2 so that it can subsequently be removed from the mitochondria by the action of catalase and glutathione peroxidase, researchers have suggested that the increased enzymatic activity of MnSOD would result in more production of H2O2. This in turn would create an imbalance between H2O2 generation and metabolism that would promote oxidative stress and damage within the cell. Such an imbalance might be due to reduced activity of the enzymes contributing to H2O2 elimination, such as catalase and glutathione peroxidase.35,42 The results support the hypothesis that deterioration of the mitochondrial function and increased oxidative damage are involved in AD pathogenesis. It is important, nonetheless, to point out that other genes linked to oxidative stress and antioxidant pathways may also affect susceptibility to developing AD, and that their genetic variability should therefore be studied.
Conflict of interestThe authors have no conflict of interest to declare.
Please cite this article as: de Mendonça E, Salazar Alcalá E, Fernández-Mestre M. Papel de las variantes GSTM1, GSTT1 y MnSOD en el desarrollo de enfermedad de Alzheimer de aparición tardía y su relación con el alelo 4 de APOE. Neurología. 2016;31:535–542.
