




The term ‘progressive myoclonus epilepsy’ (PME) refers to an array of clinical entities with heterogeneous causes. They are characterised by different types of epileptic seizures (mainly myoclonic), intellectual impairment, and other clinical manifestations mainly involving the cerebellum.1,2
We present the case of a 15-year-old male whose gestation and birth were uneventful. Psychomotor development and educational level were also normal until the age of 11. He had a healthy brother 3 years older. Their parents were not consanguineous, although both were from the same village of some 1000 inhabitants.
At the age of 12, the patient began to suffer episodes of disorientation lasting a few seconds, which were interpreted as absence seizures. During the following months, he presented several generalised tonic-clonic seizures. Cranial CT yielded normal results; EEG revealed overall slowing of the background activity plus some diffuse spike-wave complexes. Intermittent light stimulation generated a photoparoxysmal response at low frequencies. Epileptic activity did not increase during stages of drowsiness.
He was initially diagnosed with juvenile myoclonus epilepsy and treated with valproic acid (1500mg per day) associated with clonazepam (40mg per day). Given the lack of response to medication, doctors added phenobarbital (90mg per day) and ethosuximide (1000mg per day) to reduce generalised seizures and absence seizures, respectively.
During the following 2 years, the patient's epilepsy progressed unfavourably with increasingly frequent seizures. His different types of epileptic seizures were classified as atypical absence seizures, multifocal cortical myoclonus, and generalised tonic-clonic seizures. Doctors also observed declining academic performance with multiple cognitive deficits mainly affecting visuospatial and literacy abilities. The patient was finally diagnosed with progressive myoclonus epilepsy based on the above symptoms.
General physical examination revealed no cutaneous stigmata (phacomatosis), visceromegalies, or retinal cherry-red spots. Neurological examination revealed bradypsychia and amnestic deficit for recent events. Cranial nerves were normal, except for horizontal nystagmus with quick phase following the direction of the gaze. There were no relevant changes in the motor system or in sensitivity. The patient presented truncal ataxia, tremor in both hands that could be increased voluntarily, dysarthria, and bilateral dysmetria (finger-to-nose test).
Analytical tests, including a haemogram, renal, liver, and thyroid profiles, copper, ceruloplasmin, creatine kinase, antineuronal antibodies, and baseline and post-exercise lactate levels, all yielded normal results. Blood and urine amino acid levels were normal. Results from the lysosomal enzyme study were also unremarkable.
Brain MRI showed moderate overall cerebral and cerebellar atrophy.
The EEG showed slow background activity with generalised epileptiform discharges manifesting as spike/polyspike wave complexes of variable duration that persisted throughout the reading. Intermittent light stimulation generated a photoparoxysmal response at low frequencies. Epileptic activity did not increase during stages of drowsiness.
Striated muscle biopsy revealed no structural changes; fibre diameters were moderately variable. The basophilic intermyofibrillar network was increased by multiples fibres forming small round basophilic deposits, which were PAS-positive according to enzymatic oxidation methods.
Axillary skin biopsies (Fig. 1A) showed rounded intensely PAS-positive formations in the epithelial cells of the apocrine glands and in the ducts of the eccrine glands. Both biopsies were compatible with Lafora disease.
 Axillary skin biopsy (PAS stain, 2×400 magnification) showing round-shaped intensely PAS-positive formations in the epithelial cells of the apocrine glands and in the ducts of the eccrine glands which correspond to Lafora bodies. (B) Heart (PAS stain, 1×200 magnification). Multiple Lafora bodies in myocytes.")
(A) Axillary skin biopsy (PAS stain, 2×400 magnification) showing round-shaped intensely PAS-positive formations in the epithelial cells of the apocrine glands and in the ducts of the eccrine glands which correspond to Lafora bodies. (B) Heart (PAS stain, 1×200 magnification). Multiple Lafora bodies in myocytes.
Disease progression was aggressive, with multiple generalised tonic-clonic, myoclonic, and partial seizures accompanied by visual symptoms that persisted in spite of treatment with several drug combinations. He lost functional abilities to the point of becoming completely disabled; nasogastric feeding was required since his frequent palatal myoclonias provoked difficulty swallowing. He presented bladder and bowel incontinence and tetraparesis, and became confined to bed and armchair. He died of aspiration pneumonia 8 years after disease onset.
Autopsy revealed typical Lafora bodies in several areas of the central nervous system (especially the thalamus and cerebellum), the liver, and the heart (Fig. 1B).
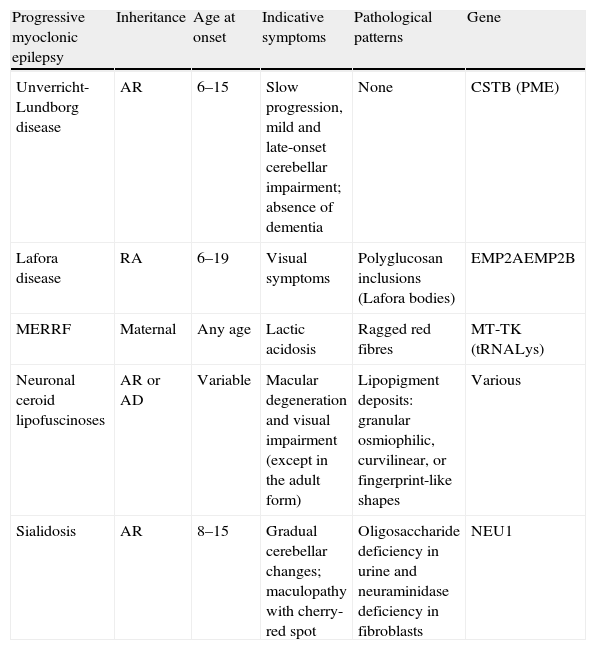
The most frequent causes of PME affecting most of the cases are Unverricht-Lundborg disease, Lafora disease, neuronal ceroid lipofuscinoses, sialidosis, and mitochondrial cytopathies (myoclonus epilepsy with ragged red fibres, MERRF) (Table 1).
Differential diagnosis of progressive myoclonic epilepsies.
| Progressive myoclonic epilepsy | Inheritance | Age at onset | Indicative symptoms | Pathological patterns | Gene |
| Unverricht-Lundborg disease | AR | 6–15 | Slow progression, mild and late-onset cerebellar impairment; absence of dementia | None | CSTB (PME) |
| Lafora disease | RA | 6–19 | Visual symptoms | Polyglucosan inclusions (Lafora bodies) | EMP2AEMP2B |
| MERRF | Maternal | Any age | Lactic acidosis | Ragged red fibres | MT-TK (tRNALys) |
| Neuronal ceroid lipofuscinoses | AR or AD | Variable | Macular degeneration and visual impairment (except in the adult form) | Lipopigment deposits: granular osmiophilic, curvilinear, or fingerprint-like shapes | Various |
| Sialidosis | AR | 8–15 | Gradual cerebellar changes; maculopathy with cherry-red spot | Oligosaccharide deficiency in urine and neuraminidase deficiency in fibroblasts | NEU1 |
AD: autosomal dominant; AR: autosomal recessive; MERRF: myoclonus epilepsy with ragged red fibres.
Dr Rodríguez Lafora was the first to describe Lafora disease in 1911.3 It consists of a degenerative and progressive disorder of the central nervous system with a recessive autosomal inheritance pattern. This disease presents no sex-related differences and it is predominantly found in southern European countries.
Lafora disease is clinically characterised by generalised tonic-clonic seizures, myoclonias, progressive mental decline, and pyramidal, extrapyramidal, and cerebellar signs.4 It appears at the end of childhood or during adolescence (6 to 20 years) and it leads to death 10 years after the onset of the first symptoms. The disease is caused by a homozygous EPM2 mutation linked to chromosome 6q23-25, which codifies tyrosine phosphatase (laforin), a protein involved in the metabolic control of glycogen.5
From a histological point of view, it is characterised by the presence of intracytoplasmic inclusion bodies in organs such as the liver, heart, and brain. They are especially common in biopsies of axillary skin.6 These bodies include glucose polymers (polyglucosan) and their presence in an axillary skin biopsy is considered nearly pathognomonic.
Myoclonias become continuous during waking hours; they are resistant to antiepileptic medication and usually associated with occipital lobe seizure. Occipital seizures are characterised by simple visual hallucinations that are sometimes complex. These hallucinations are typical of Lafora disease. The onset of myoclonias coincides with progressive deterioration of cortical function and ataxia.
On rare occasions, electroencephalographic manifestations may appear prior to symptom onset. They are characterised by increasingly frequent spike- or polyspike-wave paroxysms. Subsequently, the baseline record becomes slower and more disorganised. Paroxysms caused by intermittent light stimulation grow more frequent and gradually become continuous; photoparoxysmal response is typical at low frequencies.7 Unlike in juvenile myoclonus epilepsy, epileptic anomalies do not increase during sleep in initial stages of Lafora disease.
Brain MRI shows no relevant changes in initial and intermediate stages of the disease; final stages are characterised by cerebral and cerebellar atrophy.8
In theory, doctors can offer genetic counselling and establish a prenatal diagnosis when the genetic anomaly has been detected in a family member.9
In conclusion, doctors should assign a suspected diagnosis of Lafora disease when a young patient (in late childhood or adolescence) begins experiencing myoclonias followed by ataxia and progressive cognitive decline with no evidence of structural changes in neuroimaging tests and no metabolic changes in the analytical study.10
Please cite this article as: Jiménez Caballero PE. Epilepsia mioclónica progresiva: Descripción de un caso de enfermedad de Lafora con autopsia. Neurología. 2013;28:584–586.
