







LRRK2 mutations have traditionally been associated with a benign phenotype of Parkinson’s disease (PD). Favourable responses to deep brain stimulation (DBS) are reported in the advanced phase.
MethodsWe performed a retrospective analysis of the clinical characteristics and progression of 13 patients with LRRK2-associated PD (13 with G2019S and 1 with I1371V). Nine patients were in the advanced phase, with a mean progression time of 7.2 years before reaching this phase.
ResultsSeven patients underwent bilateral subthalamic DBS implantation, and 2 received infusion treatment. Patients with mutation G2019S responded excellently to DBS, with Unified Parkinson’s Disease Rating Scale (UPDRS) II and III scores improving by 80% at 6 months. This response was sustained over time. The patient with mutation I1371V had a severe phenotype of the disease, and presented a moderate response to DBS. Patients with advanced LRRK2-associated PD showed predominantly frontal cognitive involvement, with significant language impairment.
ConclusionsIn these patients, progression was faster in the advanced stage of the disease. We emphasise the suitability of subthalamic DBS in the management of these patients.
Las mutaciones en el gen LRRK2 se han relacionado tradicionalmente con un fenotipo benigno de la enfermedad de Parkinson (EP). En la fase avanzada, se ha descrito una respuesta favorable a la estimulación cerebral profunda (ECP).
MétodosRetrospectivamente, hemos analizado las características clínicas y la evolución de 14 pacientes con EP debida a mutaciones en LRRK2 (EP-LRRK2), 13 G2019S y 1 I1371V. Nueve de ellos, en fase avanzada, tuvieron una evolución media de 7.2 años hasta alcanzarla.
ResultadosSiete pacientes fueron intervenidos de ECP subtalámica bilateral y dos pacientes recibieron tratamiento con una terapia de infusión. Los pacientes portadores de la mutación G2019S mostraron una excelente respuesta a la ECP, con una mejoría a los 6 meses superior al 80% en las escalas UPDRS II y UPDRS III. Esta respuesta se ha mantenido en el tiempo. El paciente con la mutación I1371V mostraba un fenotipo grave de la enfermedad y su respuesta a la ECP ha sido moderada. Los pacientes con EP-LRRK2 en fase avanzada mostraron una afectación cognitiva predominantemente frontal con un deterioro significativo del lenguaje.
ConclusionesEn nuestros pacientes EP-LRRK2 hemos observado un fenotipo con una evolución más rápida a la fase avanzada de la enfermedad. Recalcamos la idoneidad de la ECP subtalámica en estos casos.
PARK8 is the most common monogenic cause of familial Parkinson’s disease (PD); it is linked to mutations in the LRRK2 gene, at locus 12p12. This gene encodes the protein dandarin, which is believed to act as a cytoplasmic kinase in protein phosphorylation. It has also been linked to vesicular and membrane transport and to protein turnover, including the lysosomal degradation pathway.1
Several missense and nonsense mutations have been described, although only 7 have been confirmed to play a role in PD pathogenesis. The G2019S mutation is thought to cause a gain of function in kinase activity, since homozygous carriers do not develop more severe disease than heterozygous carriers.2 This is the most frequent mutation worldwide, explaining over 6% of all cases of familial PD; it has also been detected in individuals with sporadic PD, although less frequently.3,4 Penetrance is variable and depends on the patient’s age. Neuropathological findings are highly variable, with the classic accumulation of Lewy bodies in most cases, but also nigral degeneration without Lewy bodies, diffuse presence of Lewy bodies, tau-positive neurofibrillary tangles, and α-synuclein– and ubiquitin-positive glial cytoplasmic inclusions.5
The phenotype of LRRK2-associated PD is generally accepted to be indistinguishable from that of idiopathic PD. Some studies have suggested that it may be a variant with a relatively benign course and slow progression.6,7 This phenotype is usually characterised by late onset and tremor, with good response to dopaminergic drugs and mild motor complications.8 Carriers of the G2019S mutation are less likely to present cognitive impairment than patients with idiopathic PD.6,9,10 However, the R1441C mutation, which originated in the Basque Country, is associated with higher rates of motor fluctuations and axial symptoms than any other mutation or than idiopathic PD.11
Deep brain stimulation (DBS) is an advanced therapy that enables the management of motor complications in PD. The most recent evidence suggests that patients with LRRK2-associated PD, particularly those carrying the G2019S mutation, achieve favourable results with subthalamic stimulation and are therefore good candidates for DBS.12
Patients and methodsWe conducted a retrospective, observational study using a prospective registry of consecutive patients with PD assessed at the movement disorders unit of Hospital Clínico Universitario de Santiago de Compostela (Spain). The study complies with the ethical principles of the World Medical Association’s Declaration of Helsinki and was approved by the research ethics committee of Santiago de Compostela (research project code 2017/350). In accordance with the good clinical practice guidelines, we requested genetic studies for patients with early-onset PD (< 50 years of age) as part of a genetic study of patients with familial PD and first-degree relatives with PD. We identified a total of 14 patients with LRRK2 mutations, all of whom had a clinical diagnosis of PD. Their baseline characteristics are summarised in Table 1.
Baseline characteristics of patients with LRRK2-associated Parkinson’s disease.
| N = 14 | |
|---|---|
| Mean age at diagnosis, years (range) | 52.9 (3-70) |
| Women (%) | 71.4% |
| First-degree relatives affected (%) | 78.6 % |
| G2019S mutation carriers (%) | 92.9 % |
| Mean PD duration, years (range) | 11.7 (4-32) |
| Mean progression time to advanced PD, years (range) (n = 9) | 7.2 (4-14) |
PD: Parkinson’s disease.
All patients but one were carriers of the G2019S mutation, a glycine to serine substitution in exon 41. The remaining patient carried the I1371V mutation, an isoleucine to valine substitution in exon 29; this mutation had previously been described as pathogenic. None of the patients carried the R1441G mutation, the most frequent mutation in the Basque Country and neighbouring regions. This may be due to geographical factors resulting in a relative isolation of this region from northern Spain, and supports the hypothesis of a geographical gradient for this mutation.13
In advanced PD, first-line treatments no longer achieve adequate control of the disease. All patients but one underwent neuropsychological evaluation. Overall cognitive status was assessed with the Mattis Dementia Rating Scale (MDRS). Memory was assessed with the TAVEC test (Test de Aprendizaje Verbal España-Complutense),14 which includes subscales for immediate recall (TAVEC-IR), short-term free recall (TAVEC-STFR), long-term free recall (TAVEC-LTFR), and memory retrieval (TAVEC-MR). The Frontal Assessment Battery (FAB) and the 4-disc Tower of Hanoi were used to assess executive function. Working memory was evaluated with the digit span test from the Wechsler Adult Intelligence Scale (WAIS). Patients also performed phonemic and semantic verbal fluency tasks (words beginning with ‘p’ and animals). The Boston Naming Test (BNT) was used to evaluate language and the Geriatric Depression Scale to detect depression.
ResultsNine of the 14 participants had advanced PD. At present, all patients are receiving advanced therapies. We recommended DBS as the first-line treatment for advanced PD, although 2 patients did not meet the eligibility criteria.
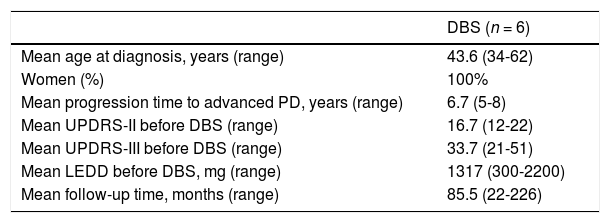
Deep brain stimulation for carriers of the G2019S mutationSix patients underwent bilateral subthalamic DBS (Table 2). Mean age at diagnosis was 43.6 years (range, 34-62) and disease duration before the advanced stage was 6.7 years (range, 5-8). Follow-up times varied considerably, with a mean of 85.5 months (range, 22-226).
Baseline characteristics of G2019S mutation carriers undergoing surgery for deep brain stimulation.
| DBS (n = 6) | |
|---|---|
| Mean age at diagnosis, years (range) | 43.6 (34-62) |
| Women (%) | 100% |
| Mean progression time to advanced PD, years (range) | 6.7 (5-8) |
| Mean UPDRS-II before DBS (range) | 16.7 (12-22) |
| Mean UPDRS-III before DBS (range) | 33.7 (21-51) |
| Mean LEDD before DBS, mg (range) | 1317 (300-2200) |
| Mean follow-up time, months (range) | 85.5 (22-226) |
DBS: deep brain stimulation; LEDD: levodopa equivalent daily dose; PD: Parkinson’s disease; UPDRS: Unified Parkinson’s Disease Rating Scale.
Response to DBS was outstanding (Fig. 1), with patients achieving an 80% decrease in UPDRS-II scores (ON medication/ON stimulation) and an 83% decrease in UPDRS-III scores (OFF medication/ON stimulation) at 6 months with respect to the latest assessment during OFF time (16.7 vs 3.3 points for the UPDRS-II, and 33.7 vs 5.7 points for the UPDRS-III). The levodopa-equivalent daily dose (LEDD) was reduced by more than half (from 1317 mg to 563 mg).

Follow-up of the carriers of the G2019S mutation undergoing deep brain stimulation.
DBS: deep brain stimulation; LEDD: levodopa equivalent daily dose; UPDRS-II: Unified Parkinson’s Disease Rating Scale - activities of daily living; UPDRS-III: Unified Parkinson’s Disease Rating Scale - motor subscale.
This response remained unchanged over the first years after DBS. Five patients were followed up for over 4 years; despite the inevitable worsening, improvements remained reasonably stable (48% on UPDRS-II and 66% on UPDRS-III). At 48 months, the mean LEDD was even lower (515 mg), which reflects the good clinical control achieved with DBS.
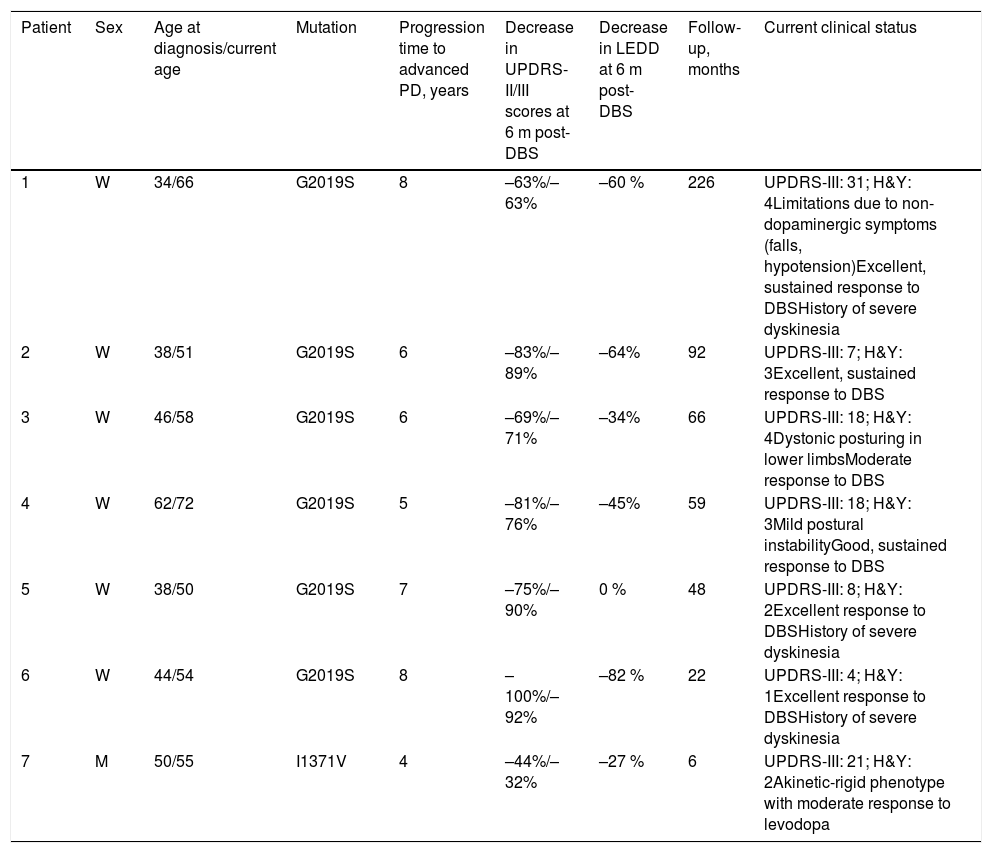
Long-term progression was satisfactory overall, although some patients presented complications associated with the course of the disease (Table 3). A good example is patient 1, a woman who had undergone surgery 19 years previously. The patient presented severe dyskinesias and was initially selected for pallidotomy; however, with the emergence of DBS around that time, her treatment was delayed until the year 2000. She continues to show good response in terms of dopaminergic symptoms, although she also presents non-dopaminergic symptoms (gait alterations, postural instability, and orthostatic hypotension). Patient 3 initially displayed good response to DBS; however, she currently presents dystonic posturing of the lower limbs, despite undergoing surgery for a second time.
Current clinical status of the patients with LRRK2-associated Parkinson’s disease undergoing deep brain stimulation.
| Patient | Sex | Age at diagnosis/current age | Mutation | Progression time to advanced PD, years | Decrease in UPDRS-II/III scores at 6 m post-DBS | Decrease in LEDD at 6 m post-DBS | Follow-up, months | Current clinical status |
|---|---|---|---|---|---|---|---|---|
| 1 | W | 34/66 | G2019S | 8 | –63%/–63% | –60 % | 226 | UPDRS-III: 31; H&Y: 4Limitations due to non-dopaminergic symptoms (falls, hypotension)Excellent, sustained response to DBSHistory of severe dyskinesia |
| 2 | W | 38/51 | G2019S | 6 | –83%/–89% | –64% | 92 | UPDRS-III: 7; H&Y: 3Excellent, sustained response to DBS |
| 3 | W | 46/58 | G2019S | 6 | –69%/–71% | –34% | 66 | UPDRS-III: 18; H&Y: 4Dystonic posturing in lower limbsModerate response to DBS |
| 4 | W | 62/72 | G2019S | 5 | –81%/–76% | –45% | 59 | UPDRS-III: 18; H&Y: 3Mild postural instabilityGood, sustained response to DBS |
| 5 | W | 38/50 | G2019S | 7 | –75%/–90% | 0 % | 48 | UPDRS-III: 8; H&Y: 2Excellent response to DBSHistory of severe dyskinesia |
| 6 | W | 44/54 | G2019S | 8 | –100%/–92% | –82 % | 22 | UPDRS-III: 4; H&Y: 1Excellent response to DBSHistory of severe dyskinesia |
| 7 | M | 50/55 | I1371V | 4 | –44%/–32% | –27 % | 6 | UPDRS-III: 21; H&Y: 2Akinetic-rigid phenotype with moderate response to levodopa |
DBS: deep brain stimulation; M: man; W: woman; H&Y: Hoehn and Yahr Scale; LEDD: levodopa equivalent daily dose; PD: Parkinson’s disease; UPDRS-II: Unified Parkinson’s Disease Rating Scale - activities of daily living; UPDRS-III: Unified Parkinson’s Disease Rating Scale - motor subscale.
The patient carrying the I1371V mutation (patient 7) presented a particularly aggressive disease course (Table 3). He was diagnosed with Parkinson’s disease at the age of 50 years, and presented olfactory and gustatory alterations, as well as sudden movements during sleep. He progressed to advanced PD within 4 years, receiving high doses of dopaminergic drugs, with a moderate response. Despite the improvements achieved with DBS, this patient presented the poorest results at 6 months of treatment. Furthermore, this patient achieved less marked decreases in UPDRS-II and UPDRS-III scores (44% and 32%, respectively) and in the LEDD (27%) than other patients in our sample.
Infusion therapiesTwo patients with advanced LRRK2-associated PD did not meet the eligibility criteria for DBS and were therefore started on infusion therapy.
One of them, a 77-year-old woman with a 14-year history of PD, was started on continuous apomorphine infusion. She has been receiving this treatment for 6 years, showing a sustained response with no serious adverse reactions. Despite her advanced age, she receives continuous apomorphine infusion for 23 hours per day, with a total dose of 120 mg per day.
The other patient, a man with a disease progression time of 7 years, was initially scheduled for DBS, but was subsequently found to have amnestic mild cognitive impairment and significant language impairment. He was therefore treated with continuous intrajejunal infusion of levodopa-carbidopa intestinal gel (DUO), showing an adequate response after 16 months of treatment. The LEDD currently used is relatively high (1700 mg), but well tolerated.
Neuropsychological assessmentEight patients underwent neuropsychological assessment. However, neuropsychological test results were not available for patient 1, as she underwent DBS implantation over 19 years ago. Table 4 shows mean scores in the tests administered. Fig. 2 presents the age- and education-adjusted neuropsychological profile of our sample, based on the standard deviation (SD) for each test in a normally distributed population (z-score). Test results are considered normal if they are higher than 1 standard deviation below the mean.
Neuropsychological assessment of patients with advanced LRRK2-associated Parkinson’s disease (n = 8).
| MDRS |
| TAVEC-IR |
| TAVEC-STFR |
| TAVEC-LTFR |
| TAVEC-MR |
| FAB |
| 4-Disc Tower of Hanoi |
| WAIS (digit span) |
| Phonemic fluency |
| Semantic fluency |
| BNT (n = 6) |
| GDS |
Data are expressed as either mean ± SD (range) or number (%).
BNT: Boston Naming Test; FAB: Frontal Assessment Battery; GDS: Geriatric Depression Scale; IR: immediate recall; LTFR: long-term free recall; MDRS: Mattis Dementia Rating Scale; MR: memory retrieval; STFR: short-term free recall; TAVEC: Test de Aprendizaje Verbal España-Complutense; WAIS: Wechsler Adult Intelligence Scale.

Neuropsychological profile of patients with advanced LRRK2-associated Parkinson’s disease.
BNT: Boston Naming Test; FAB: Frontal Assessment Battery; LTFR: long-term free recall; MDRS: Mattis Dementia Rating Scale; MR: memory retrieval; PF: phonemic fluency; STFR: short-term free recall; TAVEC: Test de Aprendizaje Verbal España-Complutense; WAIS: Wechsler Adult Intelligence Scale.
Global cognitive function in our sample was normal, with a mean (SD) MDRS score of 130.4 (9.2) points. Patients showed predominantly frontal cognitive dysfunction, with relatively preserved memory. Patients scored lowest on the FAB (14.4 [2.0]; Z = –2.26 [1.53]). Scores were normal on tasks assessing such frontal lobe functions as working memory (WAIS-digit span: 11.1 [4.3]; Z = –0.44 [1.08]) and phonemic verbal fluency (13.3 [6.4]; Z = –0.34 [1.02]). Despite being low, mean TAVEC-STFR (9.5 [4.3]; Z = –0.44 [1.08]), TAVEC-LTFR (13.3 [6.4]; Z = –0.34 [1.02]), and TAVEC-MR scores (13.6 [2.4]; Z = 0.0 [0.76]) were within the normal range. BNT scores showed considerable language impairment (42.5 [13.7]; Z = –2.62 [1.99]).
Interestingly, the patient receiving DUO scored the lowest on the MDRS, TAVEC, and BNT, but not in tasks assessing frontal lobe function; this is the main reason why subthalamic DBS was ruled out in his case.
None of the patients presented severe depression, and only 3 presented Geriatric Depression Scale scores suggestive of depression. Patient 6 presented an impulse control disorder (gambling), which was controlled by reducing the dosage of the dopamine agonist, and especially after DBS implantation.
DiscussionOur patients with LRRK2 mutations, most of whom were carriers of the G2019S mutation, presented advanced PD relatively early. The 9 patients with advanced PD reached this stage of the disease after a mean of 7.2 years of progression. In the patients undergoing DBS, progression times were even shorter (6.7 years). Only one of the 5 patients who did not have advanced PD presented a disease progression time longer than 10 years.
Progression time to advanced PD was considerably shorter in our sample than in recently published series. In the EuroInf 2 study,15 mean time to onset of advanced therapy was longer than 10 years of progression for DBS, and longer than 13 years for infusion therapies. Before onset of an advanced therapy, patients presented the following motor scores (UPDRS-III and Hoehn and Yahr Scale, respectively) during ON states: 24.1 (11.3) and 2.5 for patients undergoing DBS, 29.2 (11.0) and 3.0 for patients receiving APO, and 29.9 (12.2) and 3.0 for patients receiving DUO. In our view, these scores reveal advanced PD. In these series, infusion therapies were started in some patients at Hoehn and Yahr stages of 4.0, indicating presence of prominent axial symptoms.
In a prospective study of patients undergoing DBS, the authors observed that patients with LRRK2-associated PD developed more levodopa-induced complications and presented a significantly shorter “honeymoon period” than patients with idiopathic PD (3.8 [1.7] vs 5.4 [2.6] years).16 In another study including a large cohort of patients with PD from North Africa, nearly 40% of whom carried LRRK2 mutations, patients with LRRK2-associated PD showed a similar clinical profile to those with idiopathic PD, although they were more likely to present motor fluctuations and dyskinesias.17 These findings are consistent with our results, as they suggest that the advanced stage of PD may occur earlier than previously thought.
The outcome of DBS is linked to patient-related factors and the surgical technique used. Younger age, good response to levodopa, and absence of significant cognitive impairment or refractory axial symptoms are key to achieving a good prognosis. Correct placement of the electrode in the target region, stimulation parameters, and treatment adjustments after surgery are also essential for a good outcome. Monogenic forms of PD are associated with certain clinical phenotypes; however, little is known about the mechanisms by which these genetic factors influence the outcomes of DBS. Nearly all patients with LRRK2-associated PD displayed an outstanding (> 50%) or satisfactory response (30%-50%) to subthalamic DBS in the short (2 years) and medium term (2-6 years), except in patients with the R1441G mutation.12 In the case of the G2019S mutation, the most widely known, no significant differences have been found between carriers and controls for motor assessment results, LEDD reduction, cognitive assessment results, or neuropsychiatric disorders.18–21 Sayad et al.16 reported better motor results after DBS in patients with LRRK2-associated PD than controls with idiopathic PD; however, this may be explained by the fact that the control group did not achieve the normal improvement. Our findings are consistent with the available evidence. To our knowledge, patient 1 from our sample presents the longest follow-up period reported to date for a patient with LRRK2-associated PD undergoing DBS implantation.
Little is known about the clinical phenotype associated with the I1371V mutation. This mutation has been found to play a pathogenic role in familial PD, with carriers presenting classical PD.22,23 A post-mortem study identified typical findings of PD in a patient with late-onset parkinsonism and cognitive impairment24 and another study revealed olivopontocerebellar degeneration in a patient with clinical diagnosis of multiple system atrophy.25 To our knowledge, our patient with the I1371V mutation is the first to be treated with DBS.
Belarbi et al.26 performed an extensive neuropsychological and neuropsychiatric assessment of patients with LRRK2-associated PD, finding no statistically significant differences with respect to non-carriers. Neuropsychological assessment results were expressed as the percentage of patients with abnormal results, which hinders comparison with our own results. Abnormal results on memory tasks, the FAB, and verbal fluency tasks were more frequent among patients with LRRK2-associated PD. Our neuropsychological assessment reveals that patients with LRRK2-associated PD have a global cognitive profile typical of moderate-to-advanced PD, with predominantly frontal involvement but no significant memory impairment, and language impairment. As far as we know, no similar results have been reported. Only 6 patients were evaluated with the BNT, preventing us from extrapolating our findings. It would be interesting to compare these results against data from patients with advanced idiopathic PD undergoing DBS, or to evaluate language impairment in patients in the early stages of LRRK2-associated PD.
Our study has several limitations, including its retrospective design and the heterogeneity of the sample. However, the main limitation is the fact that genetic studies are commonly performed in younger patients, who are in turn more likely to develop motor complications, and are therefore eligible for advanced therapies, such as DBS. This may have hindered the detection of patients with a more benign disease course; thus, we are unable to determine whether the prevalence of advanced PD is higher in patients with LRRK2-associated PD.
In view of the growing interest in monogenic forms of PD, and in the context of research into specific clinical subtypes of the disease and treatments targeting specific pathogenic mechanisms, we may conclude that the course of PD may be more aggressive in patients with LRRK2 mutations. However, our patients with LRRK2-associated PD responded well to advanced therapies, particularly DBS. In our view, this may indicate that LRRK2-associated PD is a more purely motor form of the disease.
Conflicts of interestThe authors have no conflicts of interest to declare.
