



Diversos estudios han descrito que en los cerebros de pacientes con enfermedad de Alzheimer (EA) hay una mayor oxidación de lípidos, proteínas y ADN. Además, en estos pacientes se ha observado diferencias en la actividad y polimorfismos de los genes que codifican las enzimas GST (T1 y M1) y MnSOD. En virtud de ello se planteó estudiar la variabilidad de los genes GSTT1, GSTM1 y MnSOD en individuos venezolanos sanos y con EA.
MétodosSe incluyeron 179 individuos venezolanos, no relacionados, agrupados en pacientes con EA (n=79) e individuos sanos (n=100). La presencia o ausencia de los genes GSTT1/GSTM1 se determinó por PCR-SSP y los polimorfismo de los genes MnSOD y APOE por PCR-RFLP.
ResultadosEl genotipo GSTT1+/GSTM1− parece favorecer el desarrollo de la EA (OR=2,06; p=0,01), siendo el riesgo mayor al estar en combinación con el alelo ¿4 del gen APOE: GSTT1+/GSTM1−/¿3¿4 (OR=3,07; p=0,05), GSTT1+/GSTM1−/¿4¿4 (OR=5,52; p=0,02). El polimorfismo Ala-9Val por sí solo no parece estar relacionado con la EA, sin embargo, la presencia del genotipo Ala/Ala incrementa el riesgo que proporciona el alelo ¿4 del gen APOE: AlaAla/¿3¿4 (OR=3,47; p=0,03), AlaAla/¿4¿4 (OR=6,3; p=0,01).
ConclusionesLos resultados apoyan la hipótesis de que el deterioro de la función mitocondrial y el aumento de daño oxidativo están involucrados en la patogénesis de la EA. Es importante estudiar otros genes relacionados con estrés oxidativo y vías antioxidantes, los cuales pudiesen estar involucrados en la susceptibilidad a desarrollar la EA.
Several studies have reported increased oxidation of lipids, proteins and DNA in the brains of patients with Alzheimer disease (AD). Moreover, these patients display differences in the activity and polymorphisms of the genes encoding the enzymes GST (T1, M1) and MnSOD. For these reasons, we designed a study of the variability in GSTT1, GSTM1, and MnSOD genes in healthy and AD groups from a Venezuelan population.
MethodsWe included 179 unrelated Venezuelan subjects classified as either AD patients (n=79) or healthy individuals (n=100). Presence or absence of the GSTT1/GSTM1 genes was determined using PCR-SSP, and polymorphisms of MnSOD and APOE genes were identified with PCR-RFLP.
ResultsThe genotype GSTT1+/GSTM1− seems to favour development of AD (OR=2.06, P=.01). The risk level is higher when it is combined with the ¿4 allele of the APOE gene: GSTT1+/GSTM1−/¿3¿4 (OR=3.07, P=.05), GSTT1+/GSTM1−/¿4¿4 (OR=5.52, P=.02). The Ala-9Val polymorphism does not appear to be related to AD. However, the presence of the Ala/Ala genotype increases the risk provided by the ¿4 allele of the APOE gene: AlaAla/¿3¿4 (OR=3.47, P=.03), AlaAla/¿4¿4 (OR=6.3, P=.01).
ConclusionsThe results support the hypothesis that impaired mitochondrial function and increased oxidative damage are involved in the pathogenesis of AD. It is important to study other genes related to oxidative stress and antioxidant pathways which could be involved in susceptibility to AD.
En las enfermedades neurodegenerativas, como la enfermedad de Alzheimer (EA), el daño oxidativo es común, aunque no está claro si es causa o consecuencia de la enfermedad1. El daño oxidativo puede ser causado por el péptido β amiloide (Aβ) y los filamentos pareados helicoidalmente de la proteína tau, los cuales constituyen las lesiones características de la EA. Se ha descrito que el péptido Aβ puede originar radicales libres2 e inhibir la enzima citocromo oxidasa, contribuyendo así al estrés oxidativo3. Igualmente, se ha evidenciado que los filamentos pareados helicoidalmente de la proteína tau pueden formar productos avanzados de glucación, generando especies reactivas de oxígeno (ERO) suficientes para causar daño oxidativo4. Al poseer el cerebro un alto consumo de oxígeno, con altas demandas de energía, así como una capacidad antioxidante limitada en comparación con otros tejidos, lo hacen muy susceptible al daño oxidativo, por lo que es de gran importancia que las defensas antioxidantes sean efectivas en la eliminación de los radicales libres5. En pacientes con EA se han observado alteraciones en la actividad de las enzimas glutatión S transferasa (GST) y superóxido dismutasa de manganeso (MnSOD)6. Las GST constituyen una familia de enzimas que ejercen un control crítico en la protección celular contra sustancias tóxicas y estrés oxidativo y son codificadas por, aproximadamente, 16 genes, subdivididos en 8 clases7. La clase μ comprende 5 isoenzimas diferentes, denominadas desde GSTM1 a GSTM5, mientras que la clase θ comprende 2 isoenzimas, la GSTT1 y la GSTT2. Los genotipos nulos de los genes GSTM1 y GSTT1 se caracterizan por una eliminación homocigota del gen completo, por lo que no hay actividad de la enzima (revisado en Cooper8). Tanto la enzima GSTT1 como la GSTM1 son conocidas por su capacidad de catalizar la desintoxicación de oxígeno reactivo y los productos de la peroxidación lipídica9, por esta razón la inactividad enzimática de GSTT1/M1 se relaciona con una mayor exposición al estrés oxidativo10.
La enzima superóxido dismutasa (SOD) es una de las principales defensas contra los daños que puede provocar el radical superóxido (O−2), catalizando la conversión del superóxido en oxígeno molecular (O2) y peróxido de hidrógeno (H2O2), que luego será convertido en agua por la acción de la enzima catalasa o la glutatión peroxidasa11. Existen 3 isoformas de esta enzima: la citosólica [Cu/ZnSOD], la extracelular [EC-SOD] y la mitocondrial [MnSOD]12. El 90% de las ERO se originan en la mitocondria, por lo que la MnSOD es un antioxidante crítico en la protección de las células frente al estrés oxidativo. La actividad de esta enzima mitocondrial provee una defensa frente a la peroxidación de lípidos y, a nivel cerebral, protege la viabilidad de la membrana neuronal13. La enzima MnSOD es sintetizada en el citosol y luego es transportada a la mitocondria. En este transporte está involucrada una secuencia de 24 aminoácidos que se denomina secuencia blanco mitocondrial (MTS), la cual forma una estructura anfifílica helicoidal necesaria para su transporte hacia la mitocondria14. Se ha descrito que el gen que codifica para la MnSOD posee un polimorfismo que consiste en una sustitución de una timina (T) por una citosina (C) en el nucleótido 47, provocando un cambio de aminoácido (Val →Ala) en la posición −9 de la MTS, causando alteraciones en la estructura de la enzima. Estos cambios en la estructura afectan el transporte de la MnSOD dentro de la mitocondria y, por tanto, afectan la defensa celular contra los radicales superóxidos14. Asimismo, existen diversos estudios que muestran el papel de la MnSOD en la supervivencia de las neuronas frente al estrés oxidativo15,16. Considerando el papel de las enzimas GSTT1, GSTM1 y MnSOD en la defensa frente al estrés oxidativo y la existencia de una posible relación entre el estrés oxidativo y la patogénesis de la EA, nos planteamos estudiar el papel de los polimorfismo de los genes GSTT1, GSTM1 y MnSOD en el desarrollo de dicha enfermedad y correlacionar las asociaciones con la presencia del alelo ¿4 del gen APOE.
Materiales y métodosSujetosEl estudio fue realizado en 79 pacientes (edad promedio de 70±10años) diagnosticados con EA del tipo esporádico, quienes acudieron al Servicio de Neurología del Hospital Clínico Universitario de Caracas, entre septiembre del 2004 y octubre del 2006. Estos pacientes fueron seleccionados según el protocolo clínico aplicado en la Unidad de Neuropsicología Dr. Luis Borges, del Servicio de Neurología del Hospital Clínico Universitario de Caracas. Dicho protocolo se estructuró de acuerdo a los criterios de la Sociedad Psiquiátrica Americana (DSM-IV) del Instituto Nacional de Trastornos Neurológicos, de la Comunicación y de Accidentes Vasculares Cerebrales (NINCDS) y los criterios de la Asociación para la Enfermedad de Alzheimer y Trastornos Conexos (ADRDA).
El grupo control estuvo conformado por 100 individuos sanos, venezolanos, con un promedio de edad de 71±10años. A este grupo se le realizó el miniexamen mental de Folstein, las pruebas de laboratorio e imagenológicas.
Todos los individuos participantes en el estudio firmaron un consentimiento informado, el cual en el caso de los pacientes con EA fue autorizado por su representante. Dicho consentimiento fue aprobado por el Comité de Bioética del IVIC y del Hospital Clínico Universitario de Caracas.
Extracción del ADN genómicoEl ADN genómico fue extraído de los leucocitos y linfocitos de sangre periférica siguiendo el método de Bunce17.
Detección de los genes GSTT1/GSTM1Para detectar la presencia o ausencia de los genes GSTM1 y GSTT1 se utilizó un protocolo de PCR múltiplex, siguiendo la metodología descrita por Lin et al.18, con algunas modificaciones, y los iniciadores específicos publicados por Chen et al.19.
Genotipificación del gen MnSODEl polimorfismo Ala-9Val del gen MnSOD se estudió mediante la técnica de PCR-RFLP, utilizando los iniciadores y el protocolo descrito por Ambrosone et al.20.
Genotipificación del gen APOELa genotipificación y evaluación del polimorfismo de APOE se realizó mediante PCR-RFLP, utilizando los iniciadores descritos por Emi et al.21 y utilizando el protocolo descrito por Hixson y Vernier22.
Análisis estadísticosLas frecuencias alélicas y genotípicas fueron calculadas. La significación estadística de las diferencias de frecuencias (alelos, genotipos, combinaciones genotípicas) entre los grupos fue estimada por la prueba de ji cuadrado (X2) Mantel-Haenszel usando tablas de contingencia 2×2. Los valores de p se corrigieron multiplicándolos por el número de comparaciones hechas (corrección de Bonferroni) y se consideraron significativas cuando el valor de p<0,05.
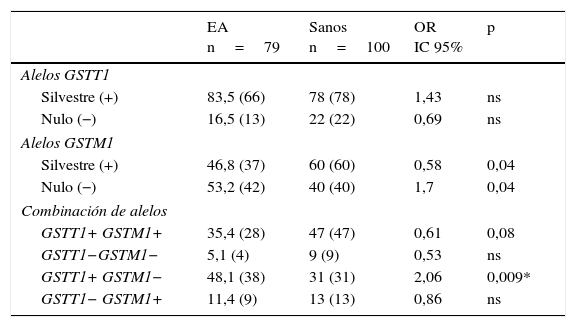
ResultadosDistribución de los genotipos y combinación genotípica de los genes GSTT1-GSTM1 en individuos sanos y pacientes con enfermedad de AlzheimerEn la tabla 1 se muestra la distribución de los genotipos GSTM1 [silvestre (+), nulo (−)] y GSTT1 [silvestre (+), nulo (−)] en individuos sanos y pacientes con EA. Al comparar las frecuencias, el genotipo GSTM1 silvestre presentó una frecuencia significativamente incrementada en los sanos con respecto a los pacientes (OR: 0,58; IC 95%: 0,3235-1,0659; p=0,04; pc=ns). En contraste, el genotipo GSTM1 nulo presentó una frecuencia significativamente incrementada en los pacientes con respecto a los sanos (OR: 1,7; IC 95%: 0,9381-3,0903; p=0,04; pc=ns). Sin embargo al corregir el valor de p estas perdieron significación.
Distribución de los genotipos y de las combinaciones genotípicas de los genes GSTT1 y GSTM1 en pacientes con EA e individuos sanos
| EA n=79 | Sanos n=100 | OR IC 95% | p | |
|---|---|---|---|---|
| Alelos GSTT1 | ||||
| Silvestre (+) | 83,5 (66) | 78 (78) | 1,43 | ns |
| Nulo (−) | 16,5 (13) | 22 (22) | 0,69 | ns |
| Alelos GSTM1 | ||||
| Silvestre (+) | 46,8 (37) | 60 (60) | 0,58 | 0,04 |
| Nulo (−) | 53,2 (42) | 40 (40) | 1,7 | 0,04 |
| Combinación de alelos | ||||
| GSTT1+ GSTM1+ | 35,4 (28) | 47 (47) | 0,61 | 0,08 |
| GSTT1−GSTM1− | 5,1 (4) | 9 (9) | 0,53 | ns |
| GSTT1+ GSTM1− | 48,1 (38) | 31 (31) | 2,06 | 0,009* |
| GSTT1− GSTM1+ | 11,4 (9) | 13 (13) | 0,86 | ns |
EA: enfermedad de Alzheimer; IC 95%: intervalo de confianza; ns: no significativo; OR: odds ratio.
Los valores mostrados en paréntesis representan el número de individuos portadores del genotipo para el sitio polimórfico estudiado. La frecuencia está expresada en porcentaje.
* significativo después de la corrección de Bonferroni
Al realizar la combinación de los genotipos de los genes GSTT1 y GSTM1, se observaron en ambos grupos todas las combinaciones posibles. En los pacientes con EA la combinación GSTT1+/GSTM1− presentó la mayor frecuencia, seguida por las combinaciones GSTT1+/GSTM1+, GSTT1−/GSTM1+ y GSTT1−/GSTM1−. En el grupo control (sanos) la combinación GSTT1+/GSTM1+ presentó la frecuencia más elevada, seguida por las combinaciones GSTT1+/GSTM1−, GSTT1−/GSTM1+ y GSTT1−/GSTM1−. Al comparar las frecuencias, la combinación GSTT1+/GSTM1− presentó una frecuencia significativamente incrementada en los pacientes con respecto a los sanos (OR: 2,06; IC 95%: 1,1118-3,8036; p=0,009; pc=0,036) y al corregir el valor de p esta permaneció significativa (tabla 1).
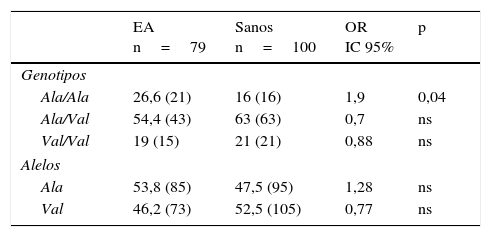
Distribución de las frecuencias alélicas y genotípicas del gen MnSOD en individuos sanos y pacientes con enfermedad de AlzheimerEn los 2 grupos el genotipo Ala/Val presentó la mayor frecuencia, seguido por los genotipos Ala/Ala y Val/Val en los pacientes y por los genotipos Val/Val y Ala/Ala en los sanos. Al compararse las frecuencias, el genotipo Ala/Va presentó una frecuencia significativamente incrementada en los pacientes con respecto a los sanos (OR: 1,9; IC 95%: 0,9147-3,9498; p=0,04; pc=ns). Sin embargo, al corregir el valor de p esta perdió significación (tabla 2).
Frecuencia alélica y genotípica del polimorfismo Ala-9Val del gen MnSOD en pacientes con EA y controles sanos
| EA n=79 | Sanos n=100 | OR IC 95% | p | |
|---|---|---|---|---|
| Genotipos | ||||
| Ala/Ala | 26,6 (21) | 16 (16) | 1,9 | 0,04 |
| Ala/Val | 54,4 (43) | 63 (63) | 0,7 | ns |
| Val/Val | 19 (15) | 21 (21) | 0,88 | ns |
| Alelos | ||||
| Ala | 53,8 (85) | 47,5 (95) | 1,28 | ns |
| Val | 46,2 (73) | 52,5 (105) | 0,77 | ns |
EA: enfermedad de Alzheimer; IC 95%: intervalo de confianza; ns: no significativo; OR: odds ratio.
Los valores mostrados en paréntesis representan el número de veces que se repite el alelo o el número de individuos portadores del genotipo para el sitio polimórfico estudiado. La frecuencia está expresada en porcentaje.
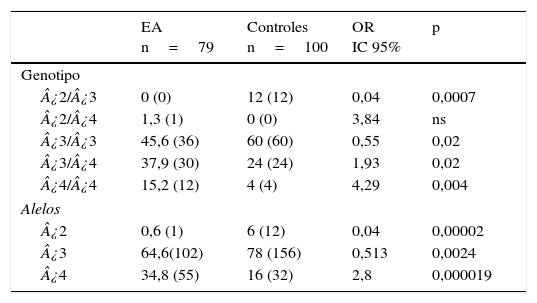
En la tabla 3 se muestran los genotipos del gen APOE presentes en pacientes y sanos. Las frecuencias de los genotipos ¿2¿3 (OR=0,04; IC 95%: 0,002-0,764; p=0,0007; pc=0,0035) y ¿3¿3 (OR=0,55; IC 95%: 0,3073-1,0136; p=0,02; pc=ns) están significativamente incrementadas en los sanos con respecto a los pacientes. En contraste, en los pacientes las frecuencias de los genotipos ¿3¿4 (OR=1,93; IC 95%: 1,0164-3,6980; p= 0,02; pc=ns) y ¿4¿4 (OR=4,29; IC 95%: 1,3241-13,9016; p=0,004; pc=0,02) están significativamente incrementadas en comparación con los sanos. Asimismo, al comparar las frecuencia alélicas, el alelo ¿2 (OR=0,04; IC 95%: 0,0059-0,3651; p=0,000025; pc= 0,000075) y el alelo ¿3 (OR=0,5; IC 95%: 0,3220-0,8195; p=0,0024; pc=0,0072) presentaron una frecuencia significativamente incrementada en los sanos con respecto a los pacientes. No obstante, en los pacientes el alelo ¿4 presentó una frecuencia significativamente incrementada con respecto a los sanos (OR=2,8; IC 95%: 1,7003-4,6221; p=0,000019; pc=0,000057).
Frecuencias de los genotipos y alelos del gen APOE en pacientes con EA e individuos sanos
| EA n=79 | Controles n=100 | OR IC 95% | p | |
|---|---|---|---|---|
| Genotipo | ||||
| ¿2/¿3 | 0 (0) | 12 (12) | 0,04 | 0,0007 |
| ¿2/¿4 | 1,3 (1) | 0 (0) | 3,84 | ns |
| ¿3/¿3 | 45,6 (36) | 60 (60) | 0,55 | 0,02 |
| ¿3/¿4 | 37,9 (30) | 24 (24) | 1,93 | 0,02 |
| ¿4/¿4 | 15,2 (12) | 4 (4) | 4,29 | 0,004 |
| Alelos | ||||
| ¿2 | 0,6 (1) | 6 (12) | 0,04 | 0,00002 |
| ¿3 | 64,6(102) | 78 (156) | 0,513 | 0,0024 |
| ¿4 | 34,8 (55) | 16 (32) | 2,8 | 0,000019 |
EA: enfermedad de Alzheimer; IC 95%: intervalo de confianza; ns: no significativo; OR: odds ratio.
Los valores mostrados en paréntesis representan el número de individuos portadores del genotipo. La frecuencia está expresada en porcentaje
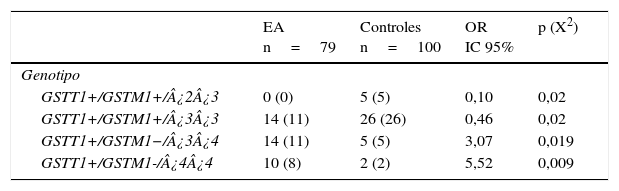
De las 24 combinaciones GSTT1/GSTM1/APOE posibles se observaron 11 en el grupo EA y 15 en sanos. Las frecuencias de las combinaciones GSTT1+/GSTM1+/¿2¿3 (OR=0,1; IC 95%: 0,0059-2,004; p=0,02; pc=ns) y GSTT1+/GSTM1+/¿3¿3 (OR=0,46; IC 95%: 0,2114-1,002; p=0,02; pc=ns) están significativamente incrementadas en sanos con respecto a los pacientes. Por el contrario, las frecuencias de las combinaciones genotípicas GSTT1+/GSTM1−/¿3¿4 (OR=3,07; IC 95%: 1,0210-9,2514; p=0,019; pc=ns) y GSTT1+/GSTM1−/¿4¿4 (OR=5,52; IC 95%: 1,1381-26,7837; p=0,009; pc=ns) están significativamente incrementadas en los pacientes con respecto a los sanos (tabla 4).
Frecuencias de las combinaciones genotípicas GSTT1/GSTM1/APOE en pacientes con EA e individuos sanos
| EA n=79 | Controles n=100 | OR IC 95% | p (X2) | |
|---|---|---|---|---|
| Genotipo | ||||
| GSTT1+/GSTM1+/¿2¿3 | 0 (0) | 5 (5) | 0,10 | 0,02 |
| GSTT1+/GSTM1+/¿3¿3 | 14 (11) | 26 (26) | 0,46 | 0,02 |
| GSTT1+/GSTM1−/¿3¿4 | 14 (11) | 5 (5) | 3,07 | 0,019 |
| GSTT1+/GSTM1-/¿4¿4 | 10 (8) | 2 (2) | 5,52 | 0,009 |
EA: enfermedad de Alzheimer; IC 95%: intervalo de confianza; OR: odds ratio; X2: prueba de ji cuadrado.
Los valores mostrados en paréntesis representan el número de individuos portadores del genotipo. La frecuencia está expresada en porcentaje
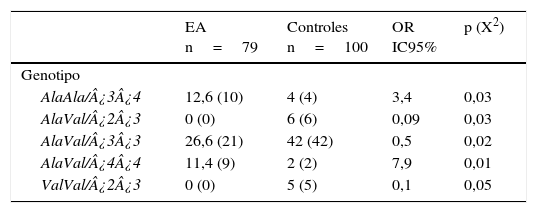
De las 18 combinaciones MnSOD/APOE posibles, 12 se observaron en sanos y 9 en pacientes. Las combinaciones AlaAla/¿3¿4 (OR=3,4; IC 95%: 1,0476-11,5486; p=0,016; pc=ns) y AlaVal/¿4¿4 (OR=7,9; IC 95%: 1,7027-36,8988; p=0,001; pc=ns) presentaron una frecuencia significativamente incrementada en pacientes con respecto a sanos. Es importante destacar que las combinaciones genotípicas AlaVal/¿2¿3 (OR=0,09; IC 95%: 0,0050-1,6480; p=0,014; pc=ns), AlaVal/¿3¿3 (OR=0,5; IC 95%: 0,2642-0,9461; p=0,016; pc=ns) y ValVal/¿2¿3 (OR=0,1; IC 95%: 0,0059-2,0049; p=0,02; pc=ns) estaban presentes únicamente en los individuos sanos (tabla 5).
Frecuencias de las combinaciones genotípicas MnSOD/APOE en pacientes con EA e individuos sanos
| EA n=79 | Controles n=100 | OR IC95% | p (X2) | |
|---|---|---|---|---|
| Genotipo | ||||
| AlaAla/¿3¿4 | 12,6 (10) | 4 (4) | 3,4 | 0,03 |
| AlaVal/¿2¿3 | 0 (0) | 6 (6) | 0,09 | 0,03 |
| AlaVal/¿3¿3 | 26,6 (21) | 42 (42) | 0,5 | 0,02 |
| AlaVal/¿4¿4 | 11,4 (9) | 2 (2) | 7,9 | 0,01 |
| ValVal/¿2¿3 | 0 (0) | 5 (5) | 0,1 | 0,05 |
EA: enfermedad de Alzheimer; IC 95%: intervalo de confianza; OR: odds ratio; X2: prueba de ji cuadrado.
Los valores mostrados en paréntesis representan el número de individuos portadores del genotipo para el sitio polimórfico estudiado. La frecuencia está expresada en porcentaje.
El desbalance entre la producción de las ERO y nitrógeno, y su eliminación es lo que se conoce como estrés oxidativo, el cual la célula debe contrarrestar para restaurar el balance redox y así evitar la pérdida de funciones neuronales y la muerte neuronal, que se han relacionado con enfermedades neurodegenerativas23. Las investigaciones relacionadas con la patogénesis de la EA se han centrado en el papel del estrés oxidativo, debido al alto consumo energético y al bajo nivel de defensa antioxidante del sistema nervioso central, que lo hacen altamente sensible al estrés oxidativo24. En el cerebro con EA los sistemas antioxidantes son menos funcionales, pudiendo guiar a un incremento de las ERO y de las especies reactivas de nitrógeno que reaccionarían con biomoléculas, incluyendo proteínas, lípidos, hidratos de carbono, ADN y ARN, provocando alteraciones en su estructura y la pérdida de sus funciones25. El estrés oxidativo en el cerebro con EA está bien documentado, observándose concentraciones alteradas de las enzimas antioxidantes, así como un aumento en la concentración de los marcadores de estrés oxidativo en el cerebro con EA en comparación con los controles pareados por edad. Además, se ha evidenciado el papel del estrés oxidativo en la progresión de la EA, al comparar la aparición de las mismas proteínas cerebrales oxidadas en sujetos con deterioro cognitivo leve, EA temprana y EA en fase tardía, mostrando que ciertas vías principales se activan y podrían estar implicadas en la progresión de la EA25. Debido a que la familia de enzimas GST conjugan el glutatión reducido con compuestos electrofílicos y de esta manera facilitan su eliminación de las células, evitando el daño oxidativo26, se han publicado estudios sobre el papel de los genes que codifican para las enzimas GSTM1 y GSTT1 en diferentes patologías. Estas enzimas poseen un fenotipo nulo, caracterizado por la ausencia de la actividad de la enzima, debido a la herencia de una eliminación homocigota del gen completo. En este estudio, el genotipo GSTM1 nulo presentó una frecuencia significativamente incrementada en los pacientes con EA (OR: 1,7; p= 0,04; pc=ns) y el genotipo GSTM1 silvestre una frecuencia significativamente incrementada en los sanos (OR: 0,58; p=0,04; pc=ns), contrastando con lo descrito en otros estudios9,27–30. Asimismo, la combinación GSTT1+/GSTM1− es más frecuente en los pacientes con EA (OR: 2,06; pc=0,036) con respecto a los sanos. Estos resultados sugieren que el genotipo GSTM1 nulo y la combinación GSTT1+/GSTM1− estarían confiriendo hasta 2 veces mayor riesgo de padecer EA en quienes lo portan. Considerando que una gran variedad de estudios sugieren que la variabilidad del gen APOE es un factor genético de riesgo en la EA31–33, se establecieron las combinaciones entre los genotipos GSTT1/GSTM1 y APOE, obteniéndose resultados interesantes. Las frecuencias de las combinaciones genotípicas GSTT1+/GSTM1−/¿3¿4 y GSTT1+/GSTM1−/¿4¿4 estaban significativamente incrementadas en los pacientes con respecto a los individuos sanos, mostrando que la presencia de las mismas podría conferir susceptibilidad al desarrollo de EA y que el riesgo que parece conferir la presencia de uno o 2 alelos ¿4 del gen APOE se incrementa en aquella combinación donde el gen GSTM1 está ausente. Por lo tanto, un aumento en la agregación del péptido Aβ debido a la isoforma E4 de la apolipoproteína E34, junto con la disminución de defensas antioxidantes, como consecuencia de la eliminación homocigota del gen GSTM1, podrían estar causando un mayor estrés oxidativo y, por ende, una mayor muerte neuronal, explicando así cómo el riesgo proporcionado por el alelo ¿4 del gen APOE se ve aumentando en ausencia del gen GSTM1. Además, las frecuencias de las combinaciones donde están presentes los genotipos GSTT1 y GSTM1 silvestres y la presencia de por lo menos un alelo ¿3 [GSTT1+/GSTM1+/¿2¿3 y GSTT1+/GSTM1+/¿3¿3] estaban significativamente incrementadas en los individuos sanos con respecto a los pacientes, sugiriendo que la presencia de las mismas podría conferir protección contra el desarrollo de la EA. Los péptidos Aβ son componentes de las placas seniles que inician la degeneración de las neuronas del cerebro en la EA al incrementar las ERO que pueden exceder las capacidades de defensa de la célula. Kaminsky y Kosenko35 investigaron los efectos in vivo de los péptidos Aβ sobre las enzimas antioxidantes, de fuentes enzimáticas mitocondriales y no mitocondriales, en cerebro de rata, y describieron que los péptidos Aβ aumentan las actividades enzimáticas formadoras de H2O2 e inhiben la actividad de las enzimas que consumen H2O2 en mitocondria y citosol, sugiriendo que el desequilibrio entre las actividades de enzimas generadoras y metabolizadoras de H2O2 contribuyen con el estrés oxidativo subyacente en la neurodegeneración y la muerte neuronal en la EA. Igualmente, el péptido Aβ también se ha visto involucrado en la disminución de la expresión de la citocromo c oxidasa en la mitocondria, afectando así la cadena transportadora de electrones y originando ERO3. La MnSOD es una enzima que forma parte de las defensas enzimáticas antioxidantes de la célula y es la primera línea de defensa de la células frente al anión superóxido convirtiéndolo en H2O212. Existen diversos estudios que proponen un papel de la MnSOD en la supervivencia de las neuronas frente al estrés oxidativo. Entre estas evidencias está el estudio realizado con ratones knockout para el gen MnSOD, en el cual se describió que estos ratones sucumbían poco tiempo después de nacer y presentaban neurodegeneración15,16. Igualmente, en ratones transgénicos se ha reportado que la deficiencia de MnSOD causa un aumento en las concentraciones del péptido Aβ, favoreciendo la formación de las placas neuríticas (revisado en Sompol et al.36). Sin embargo, otros estudios describen que la sobreexpresión de la enzima MnSOD protege a las neuronas del daño oxidativo37–39. En virtud de ello, se estudió un polimorfismo, Ala-9Val del gen MnSOD, que altera la estructura secundaria de la proteína y, por tanto, el transporte de la MnSOD dentro de la mitocondria, afectando la defensa celular contra los radicales superóxidos40. Al comparar las frecuencias de los genotipos Ala-9Val del gen MnSOD, el genotipo Ala/Ala presentó una frecuencia significativamente incrementada en pacientes con respecto a sanos, contrastando con lo descrito por Ventriglia et al. en población italiana13. Además, al evaluar el efecto combinado del gen MnSOD y APOE, las frecuencias de la combinaciones AlaAla/¿3¿4 (OR=3,4; p=0,03) y AlaVal/¿4¿4 (OR=7,9; p=0,01) estaban significativamente incrementadas en pacientes con respecto a sanos, sugiriendo que la presencia de las mismas pudiesen conferir susceptibilidad al desarrollo de EA. Por lo tanto, la presencia del alelo Ala, asociado con una mayor actividad de la enzima MnSOD humana, y la presencia del alelo ¿4 favorece el desarrollo de la EA. Cabe destacar que el riesgo que confieren los genotipos ¿3¿4 y ¿4¿4 (OR=1,93 y OR=4,29 respectivamente) se incrementa ∼2 veces al estar en combinación con los genotipos AlaAla y AlaVal del gen MnSOD (OR=3,4 y OR=7,9 respectivamente). Por el contrario, las combinaciones genotípicas AlaVal/¿2¿3, AlaVal/¿3¿3 y ValVal/¿2¿3 presentes únicamente en los individuos sanos, sugieren que la presencia de las mismas podrían conferir protección contra el desarrollo de la EA. Por lo tanto, la presencia del alelo ¿3, en forma homocigota o con el alelo ¿2, con una o 2 dosis del alelo Val, asociado con una actividad anormal de la enzima MnSOD40,41, protege a los individuos de desarrollar la EA. Fundamentados en que el transporte hacia la mitocondria de la forma Ala de la MnSOD es entre un 30-40% más eficiente que la forma Val14 y que esta enzima cataliza la dismutación del anión superóxido en H2O2, que luego es eliminado de la mitocondria por la acción de la catalasa y glutatión peroxidasa, se sugiere que la mayor actividad de la enzima MnSOD generaría una mayor producción de H2O2, causando un desbalance entre la generación y el metabolismo H2O2, favoreciendo el estrés y daño oxidativo de la célula, posiblemente debido a que la actividad de las enzimas involucradas con la eliminación del H2O2, como la catalasa y glutatión peroxidasa, se encuentran disminuidas35,42. Los resultados apoyan la hipótesis de que el deterioro de la función mitocondrial y el aumento de daño oxidativo están involucrados en la patogénesis de la EA. Sin embargo, es importante destacar que existen otros genes relacionados con estrés oxidativo y vías antioxidantes, que podrían estar involucrados en la susceptibilidad a desarrollar la EA, por lo tanto, sería relevante estudiar la variabilidad genética de dichos genes.
Conflicto de interesesLas autoras declaran no tener ningún conflicto de intereses.

