




Hereditary cerebellar ataxias (HCA) and hereditary spastic paraplegias (HSP) are a very heterogeneous group of genetic neurodegenerative diseases that may affect populations of any sex and at any age. The main symptoms consist of slowly progressive cerebellar ataxia and/or spastic paraparesis, which may be associated with other neurological or systemic manifestations.1 Their diagnosis is complex, and no disease-modifying treatment is available in most cases.2
Patients with this type of condition face several issues: insidious symptom onset that may lead the patient to consult even years after disease onset; occasionally, difficulty in identifying the nature of symptoms, which may lead to the patient being assessed by other specialists (traumatologists, otorhinolaryngologists, etc), which in turn delays diagnosis; the need for extensive and time-consuming batteries of diagnostic tests, which do not always lead to a definitive diagnosis (ie, a specific molecular diagnosis)3,4; and finally, after diagnosis, a progressive and incapacitating disease that, due to its genetic nature, may have implications not only for patients but also for their direct family members.
In this context, the aim of this study was to identify the main experiences and concerns related to the disease from the patient perspective. To that end, we recruited 12 consecutive patients with HSA/HSP from our hospital’s specialised unit between April and June 2018. Participants gave written informed consent to participate in a group interview (discussion group) addressing their main experiences and concerns regarding their condition. The discussion or focus group, which included 12 patients and 8 family members, met at a meeting room where a 2-hour session was held; 3 expert moderators led the session, and participants explained the concerns and needs arising over the course of the disease. The semi-structured questionnaire focused on the following settings: the patient’s environment (what does he/she feel/think/do?), the external resources available to the patient (physician, family environment, and community), and the progression stage (first symptoms, diagnosis, and progression). The moderators noted the participants’ answers as patients recounted their journey with the disease.
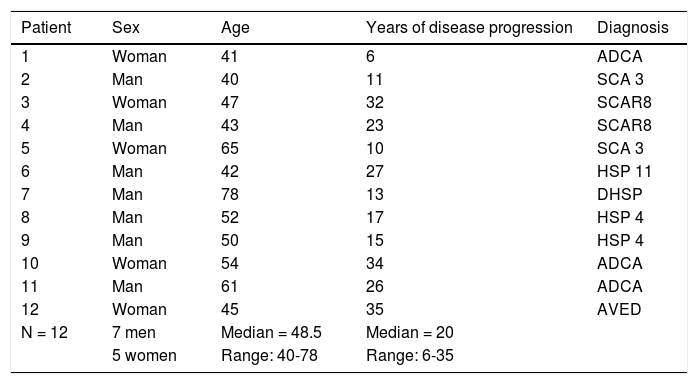
Table 1 includes the characteristics of the sample of participating patients.
Characteristics of our patient sample.
| Patient | Sex | Age | Years of disease progression | Diagnosis |
|---|---|---|---|---|
| 1 | Woman | 41 | 6 | ADCA |
| 2 | Man | 40 | 11 | SCA 3 |
| 3 | Woman | 47 | 32 | SCAR8 |
| 4 | Man | 43 | 23 | SCAR8 |
| 5 | Woman | 65 | 10 | SCA 3 |
| 6 | Man | 42 | 27 | HSP 11 |
| 7 | Man | 78 | 13 | DHSP |
| 8 | Man | 52 | 17 | HSP 4 |
| 9 | Man | 50 | 15 | HSP 4 |
| 10 | Woman | 54 | 34 | ADCA |
| 11 | Man | 61 | 26 | ADCA |
| 12 | Woman | 45 | 35 | AVED |
| N = 12 | 7 men | Median = 48.5 | Median = 20 | |
| 5 women | Range: 40-78 | Range: 6-35 |
ADCA: autosomal dominant cerebellar ataxia; AVED: ataxia with vitamin E deficiency; DHSP: dominant hereditary spastic paraplegia; HSP 4: hereditary spastic paraplegia type 4; HSP 11: hereditary spastic paraplegia type 11; SCA 3: spinocerebellar ataxia type 3; SCAR8: autosomal recessive spinocerebellar ataxia type 8.
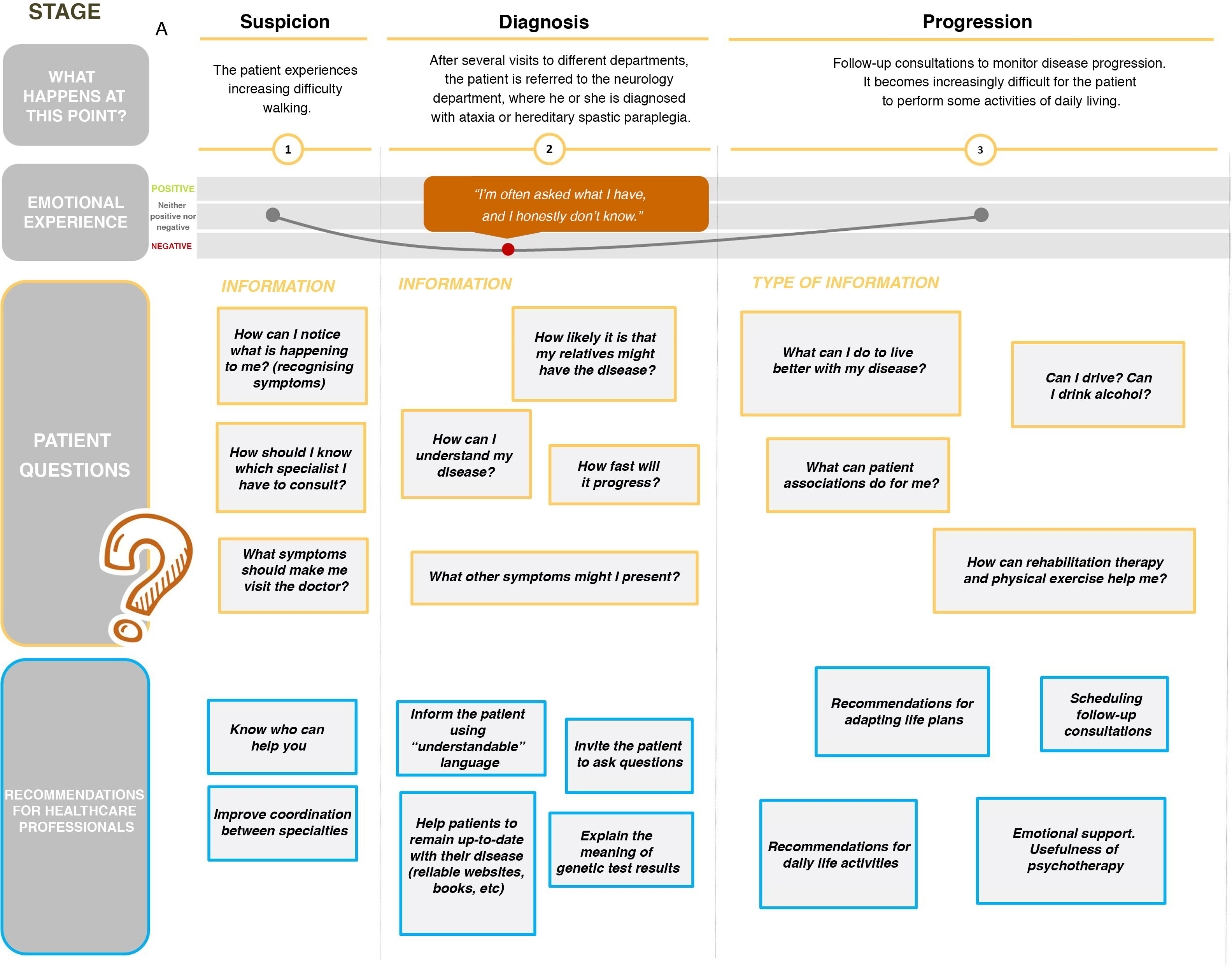
With respect to the data collected, we observed that patients’ main questions at disease onset were related to recognising the first symptoms and undergoing specialised assessment. Later, at diagnosis, patients’ emotional experience worsens, mainly due to uncertainty about the disease, its possible progression, and the consequences for their families; the need for better information on the course of the disease and the findings of the genetic tests were also mentioned. Finally, we observed an adaptation process, in which knowing such measures as the recommended lifestyle habits and the potential role of physiotherapy becomes important for the patient (Fig. 1).
Our study was conducted according to the Consolidated Criteria for Reporting Qualitative Studies (COREQ).5 Focus groups are one of the main methods in qualitative research, and constitute an essential tool for exploring the experiences of and relevant facts for a specific population group.6 Few qualitative research studies have been conducted in the field of HCA/HSP, and mainly focus on patients’ experience regarding diagnosis and the role of physiotherapy for patients with cerebellar ataxia.3,7–9 Our study provides a more global view of the main concerns and needs of patients with HCA/HSP during disease progression.
The main limitation of our study may be its retrospective approach to a disease that presents long progression times in the majority of cases (median of 20 years). Longitudinal prospective analysis with a larger sample of patients may provide more information regarding the experiences of the patients from onset and throughout follow-up, and on their relationship with healthcare services. However, the information gathered may be useful for identifying important issues for patients with HCA/HSP and their families, which may in turn be used to prepare guidelines and action protocols, and further the understanding of such subjects as specialised assessments or the role of physiotherapy and psychotherapy in HCA/HSP in future research.
Please cite this article as: Rouco Axpe I, Loyola Irulegui A, Ruiz de la Peña B, Izarzugaza Iturrizar E. Las Ataxias y Paraparesias Espásticas Hereditarias: experiencia en torno a la enfermedad desde la perspectiva del paciente. Neurología. 2021;36:736–738.
