



Information on achieving presymptomatic diagnosis of spinocerebellar ataxia (SCA) is limited. The advent of molecular diagnosis makes it possible to identify the carriers of different diseases and has also introduced the prospect of detecting diseases even before their onset. This has drawn attention to the ethical implications that must be considered in these subjects with a view to preserving their physical and psychological well-being.
DevelopmentSCA is composed of a group of neurodegenerative disorders with autosomal dominant inheritance. Only a few publications have described the genetic counselling processes and guidelines to be followed during the process of presymptomatic diagnosis (PSD). The size of the multidisciplinary teams, their areas of expertise, and the number of counselling sessions are different for each of the studies analysed here. However, the basis of presymptomatic diagnosis originates in common guidelines to which members of our team have contributed recently.
ConclusionPresymptomatic diagnosis should be performed according to guidelines that safeguard the subjects’ welfare. The diagnostic process is only recommended for patients over 18 years old with symptoms suggesting SCA, and a minimum risk of 50%. Genetic counselling programmes must be available in all centres that offer presymptomatic diagnosis of SCA.
Existe información limitada de la realización de diagnóstico presintomático en ataxias espinocerebelosas (SCA) autosómicas dominantes. La llegada del diagnóstico molecular, además de brindar la posibilidad de realizar identificación en pacientes portadores de distintas enfermedades, permitió también la posibilidad de detectar enfermedades incluso antes de su presentación. Esto atrajo la atención sobre las implicaciones éticas que deberían ser consideradas en estos sujetos, con la finalidad de salvaguardar su bienestar físico y psicológico.
DesarrolloLa SCA está compuesta por un grupo de trastornos neurodegenerativos con patrón de herencia autosómico dominante. Existen pocas publicaciones que describen el proceso de asesoramiento y los lineamientos considerados durante el proceso de diagnóstico presintomático. El número de integrantes de los equipos multidisciplinarios, sus áreas de especialidad y número de sesiones durante el asesoramiento es variable en cada uno de los trabajos analizados. Sin embargo, las bases para su realización tienen origen en documentos comunes, en los cuales algunos de los autores han participado en fechas más recientes.
ConclusionesEl diagnóstico presintomático debe ser realizado bajo lineamientos que salvaguarden el bienestar de los sujetos. Sería recomendable que el diagnóstico de SCA sea realizado solo a pacientes con clínica sugestiva, mayores de 18 años y con un riesgo mínimo del 50%. Deben estar disponibles esquemas de asesoramiento genético en todos aquellos centros que pretenden realizar diagnóstico de SCA antes de la presentación de síntomas.
With the introduction of molecular diagnosis and such programmes as the Human Genome Project, scientists expected great advances in different areas of knowledge, as well as the possibility of improving health. Primary objectives of medical genetics were clear: diagnosis, treatment, and prevention of genetic disorders.1 As a result, molecular diagnosis of such genetic disorders as Huntington disease (HD), Alzheimer disease, spinocerebellar ataxias (SCA), familial amyloid polyneuropathy, and other neurodegenerative disorders, has been possible for more than 30 years. This offered the opportunity to deliver presymptomatic diagnoses (PSD) and predictive diagnoses of some neurodegenerative diseases before any signs and symptoms are apparent.2,3 The ethical considerations of this possibility soon attracted attention, based on the controversy surrounding the benefits of ascertaining a person's susceptibility to a disease for which there is no curative treatment and whose clinical outcome cannot be modified.4,5 In the specific case of SCA, the European Molecular Genetics Quality Network (EMQN) drafted guidelines in 2010 describing requirements for performing presymptomatic analyses in accredited laboratories with the aim of providing a quality guarantee.6 However, there is no standard providing guidance on presymptomatic diagnosis in subjects at risk for presenting a disease. Currently, PSD is conducted according to general ethical guidelines and other guidelines issued for such diseases as HD.
ObjectiveThe objective is to analyse the ethical considerations, genetic counselling procedures, and recommendations derived from studies of PSD in subjects at risk for SCA.
DevelopmentPresymptomatic diagnosis is defined as the process of identifying healthy subjects who will develop a genetic disorder if they live long enough.7 It was first used in 1983 to identify subjects at risk of developing HD.8 Based on its origin and inheritance pattern, SCA was included in the group of neurodegenerative diseases that can be diagnosed before patients present symptoms.9
Spinocerebellar ataxiasSpinocerebellar ataxias are a group of neurodegenerative disorders with an autosomal dominant inheritance pattern. Their symptoms are caused by dysfunction of the cerebellum and brainstem, and of their pathways and associated connections.10,11 The incidence of SCA is estimated at 2 to 3 cases per 100 000 population.12 Ruano et al.13 performed a meta-analysis which determined a prevalence of 0 to 5.6 cases per 100 000 population for autosomal dominant SCA. Similarly, Polo et al.14 identified a prevalence of 0.29 cases per 100 000 population in Cantabria. Since the disease was first identified, different classifications have been used, with the classification based on gene loci being the most widely accepted.15 More than 35 types have been described to date.16 The subset caused by CAG repeats (SCA1, SCA2, SCA3, SCA6, SCA7, SCA12, SCA17, and DPRLA) are worth mentioning since they are responsible for more than 50% of all cases. Other SCAs originate in different types of repeats, point mutations, and deletions.17,18
On the clinical level, autosomal dominant SCAs are characterised by the presence of progressive cerebellar ataxia, which may be associated with ophthalmoplegia; pyramidal, extrapyramidal, and sensory signs; cognitive impairment; and peripheral neuropathy. Symptoms usually manifest in adults; however, several types show an anticipation phenomenon. Due to the clinical and genetic heterogeneity of this group of diseases, they should be identified by a molecular study of genomic DNA obtained from a blood sample.15 No curative treatments have been developed for this group of diseases. Current management focuses on treating symptoms and complications, in addition to appropriate genetic counselling offered by a multidisciplinary team.19,20
MethodsWith the aim of analysing the ethical considerations and genetic counselling procedures used in PSD for subjects at risk for SCA, we searched PubMed using the keywords ‘genetic counselling in SCA’ and ‘presymptomatic diagnosis and SCA’. Our search delivered 12 articles. We excluded those analysing PSD as part of a familial study, those lacking genetic counselling before the analysis, and those not clearly distinguishing between subjects at risk for SCA and those likely to develop other neurodegenerative diseases. Information was limited and only 6 studies met the inclusion criteria. We also searched for all articles cited as the basis for ethical considerations in these studies.
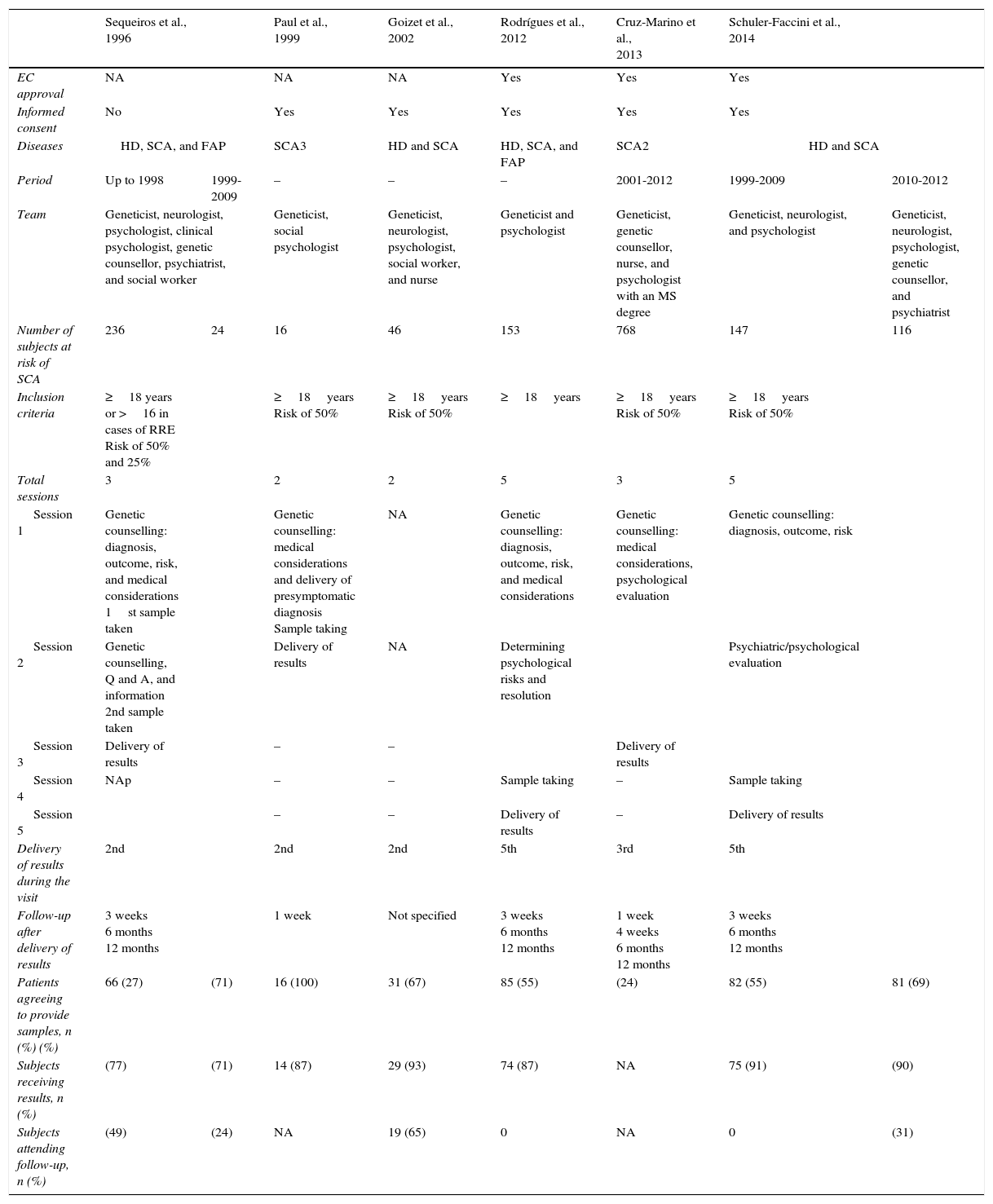
StudiesAll articles analysed in this review followed the guidelines available at that time for undertaking PSD during research. Given that other neurodegenerative disorders show similar clinical presentation and progression, some articles analysed more than one disease. Only Paul et al.21 and Cruz-Marino et al.22 included exclusively patients at risk for SCA3 and SCA2, respectively. Goizet et al.23 and Schuler-Faccini et al.24 studied subjects with risk of HD and SCA; Sequeiros et al. (1996 a,b; cited in Schuler-Faccini)24 and Rodrigues et al.8 conducted their studies in subjects at risk for HD, SCA, and familial amyloid polyneuropathy. Although different diseases were analysed in each study, there were similarities between the PSD procedures described, which makes them comparable.
One of the first points to consider in PSD is the right of any (asymptomatic) patient to remain unaware of his condition, especially when no treatment is available, since this knowledge will change the patient's self-perceived health. It may even harm patients by triggering catastrophic events, as has been observed in such diseases as HD.25 This being the case, good clinical practices in research and the guidelines issued by different organisations require an appropriate informed consent process prior to performing PSD. The consent document should highlight such important aspects as confidentiality, risks, benefits, study limitations, and others. Most of the studies analysed include signing an informed consent to be part of the genetic counselling process. In order to preserve patient confidentiality, it is important to reflect on where data are to be stored and whether they should be kept in separate files that will only be available to the interested party. Another point to consider is the maturity level of the patient receiving the information. However, each case will have to be examined individually to determine the subject's capacity. Subjects of legal age are generally thought to be prepared for this kind of decision; however, legal age differs from country to country, with a range of 14-21 years. For this reason, a consensus was reached to establish 18 years as the minimum age for participating in PSD.5 Only the study by Sequeiros et al.24 permitted an analysis of subjects aged 16 or older as part of a protocol for estimating reproductive risk. The minimum age for PSD may be a controversial issue, and analysing the subjects’ maturity levels prior to PSD is recommendable to ensure their well-being. Furthermore, every patient's right to decide freely whether they wish to know their health status should be respected. The PSD process should provide patients with the necessary tools to make the decision independently; having reached the age of 18 is used only as a reference.
Genetic counselling programmesDifferent programmes have been implemented and all of them are supported by multidisciplinary teams. Subjects must be able to receive the best quality genetic counselling and medical and psychological support to cope with PSD. With this in mind, the studies reviewed here included at least one geneticist and one psychologist. These professionals were responsible for providing information on the diagnosis, risks, prognosis, and medical details, as well as for assessing psychological well-being. Other studies also included genetic counsellors, psychiatrists, neurologists, nurses, and social workers. Table 1 presents detailed information about each study. The study by Schuler-Faccini et al. could be used to analyse how the same counselling programme was affected by changes to the multidisciplinary team, since they only included a geneticist, a psychologist, and a neurologist in the first stage. They later incorporated a genetic counsellor and a psychiatrist. This way, it is possible to compare the PSDs performed between 1999 and 2009 and those from the 2010 to 2012 period.24 There was a slight increase in the percentage of subjects willing to undergo PSD, as well as a significant increase in the percentage of patients who attended follow-up visits. No minimum number of specialties has been determined for the multidisciplinary team. In order to provide better care, the team should at the very least include a geneticist and a neurologist; these doctors will be responsible for such medical processes as diagnosis, clinical analysis, management, and treatment of the disease and its complications. Support from a psychologist, a psychiatrist, and a genetic counsellor is also necessary; these professionals will be responsible for determining the patient's maturity level, psychological integrity, and any risks derived from PSD. They also support patients as they cope with their diagnoses and accompany them during the process. However, other professionals, including nurses, social workers, and nutritionists, may also be needed to provide optimal genetic counselling services.7
Comparison of the characteristics of the studies focusing on presymptomatic diagnosis of subjects with risk of SCA.
| Sequeiros et al., 1996 | Paul et al., 1999 | Goizet et al., 2002 | Rodrígues et al., 2012 | Cruz-Marino et al., 2013 | Schuler-Faccini et al., 2014 | |||
|---|---|---|---|---|---|---|---|---|
| EC approval | NA | NA | NA | Yes | Yes | Yes | ||
| Informed consent | No | Yes | Yes | Yes | Yes | Yes | ||
| Diseases | HD, SCA, and FAP | SCA3 | HD and SCA | HD, SCA, and FAP | SCA2 | HD and SCA | ||
| Period | Up to 1998 | 1999-2009 | – | – | – | 2001-2012 | 1999-2009 | 2010-2012 |
| Team | Geneticist, neurologist, psychologist, clinical psychologist, genetic counsellor, psychiatrist, and social worker | Geneticist, social psychologist | Geneticist, neurologist, psychologist, social worker, and nurse | Geneticist and psychologist | Geneticist, genetic counsellor, nurse, and psychologist with an MS degree | Geneticist, neurologist, and psychologist | Geneticist, neurologist, psychologist, genetic counsellor, and psychiatrist | |
| Number of subjects at risk of SCA | 236 | 24 | 16 | 46 | 153 | 768 | 147 | 116 |
| Inclusion criteria | ≥18 years or >16 in cases of RRE Risk of 50% and 25% | ≥18years Risk of 50% | ≥18years Risk of 50% | ≥18years | ≥18years Risk of 50% | ≥18years Risk of 50% | ||
| Total sessions | 3 | 2 | 2 | 5 | 3 | 5 | ||
| Session 1 | Genetic counselling: diagnosis, outcome, risk, and medical considerations 1st sample taken | Genetic counselling: medical considerations and delivery of presymptomatic diagnosis Sample taking | NA | Genetic counselling: diagnosis, outcome, risk, and medical considerations | Genetic counselling: medical considerations, psychological evaluation | Genetic counselling: diagnosis, outcome, risk | ||
| Session 2 | Genetic counselling, Q and A, and information 2nd sample taken | Delivery of results | NA | Determining psychological risks and resolution | Psychiatric/psychological evaluation | |||
| Session 3 | Delivery of results | – | – | Delivery of results | ||||
| Session 4 | NAp | – | – | Sample taking | – | Sample taking | ||
| Session 5 | – | – | Delivery of results | – | Delivery of results | |||
| Delivery of results during the visit | 2nd | 2nd | 2nd | 5th | 3rd | 5th | ||
| Follow-up after delivery of results | 3 weeks 6 months 12 months | 1 week | Not specified | 3 weeks 6 months 12 months | 1 week 4 weeks 6 months 12 months | 3 weeks 6 months 12 months | ||
| Patients agreeing to provide samples, n (%) (%) | 66 (27) | (71) | 16 (100) | 31 (67) | 85 (55) | (24) | 82 (55) | 81 (69) |
| Subjects receiving results, n (%) | (77) | (71) | 14 (87) | 29 (93) | 74 (87) | NA | 75 (91) | (90) |
| Subjects attending follow-up, n (%) | (49) | (24) | NA | 19 (65) | 0 | NA | 0 | (31) |
EC, ethics committee; RRE, reproductive risk estimation; FAP, familial amyloid polyneuropathy; HD, Huntington disease; NAp, not applicable; NA, not available; SCA, spinocerebellar ataxia.
According to the available guidelines, at least 3 counselling sessions should be completed before PSD to protect subjects from risks.7 Based on the above, each study arranged for patients to have varying numbers of visits before PSD. A minimum of 2 sessions were scheduled. These offered counselling at the initial meeting, after which the sample was obtained. Results were delivered in a subsequent visit, as in the study by Goizet et al.23 Other studies such as those by Rodrigues et al.8 and Schuler-Faccini et al.24 opted for more conservative programmes which offered more in-depth care, with emphasis on analysing psychological integrity and risk. These programmes also provided the opportunity of resolving the disorders identified and subsequently taking a sample for PSD. They ended when results were delivered during the final visit. Wertz et al.7 recommended holding 3 sessions before delivering results; however, this is not a set number, and each team should adapt it specifically according to the patient's psychological integrity to minimise the risks of catastrophic events.
Similarly, follow-up sessions following presentation of the results of the PSD are also recommended. Unlimited follow-up sessions are recommended for subjects with unfavourable test results, but also for those with normal results to alleviate survivor's guilt.7 In accordance with these recommendations, the research articles we reviewed included follow-up programmes consisting of at least one visit after presenting the results with the exception of the study by Goizet et al.23, which does not mention follow-up. Most authors designed their studies to include a follow-up programme of 3 to 4 visits (Table 1), in addition to offering subsequent visits at the subjects’ request. However, the percentage of individuals reported by the studies to have attended the scheduled follow-up visits was 0%-49%, in the best case scenario. All reports suggest a low attendance rate for follow-up visits; however, no causes have been identified, except in the study by Cruz-Marino et al.22 Therefore, patients undergoing PSD testing should be informed of the importance of follow-up sessions, which afford the opportunity of remaining in contact to prevent complications and receive information on any new treatment options.
Guidelines on presymptomatic diagnosesThe guidelines issued by the World Health Organization in 2003 recommend considering the following basic conditions before offering PSD testing: (i) guarantee confidentiality, (ii) inform the subject about the limitations of testing, (iii) ensure the subject has no mental disorder when the test is performed, (iv) examine evidence that the information provided by testing would be used to prevent harm to the tested individual, families, etc., and (v) ensure that testing is accompanied by a counselling programme appropriate for the disorder. Additionally, subjects should provide their informed consent.7
In the case of SCA, requirements have been issued by such organisations as the EMQN for PSD testing in accredited laboratories. Some of the most important requirements include the following: (i) no PSD should be offered to minors since it holds no advantages for them, (ii) joint documentation of the process by the genetic counselling service and the laboratory; (iii) all requests for PSD must be made through a genetic counselling service or be accompanied by a document stating that pre-test genetic counselling took place, (iv) written informed consent according to local and/or international regulations, and (v) there must be a confirmed diagnosis in the family.6
Unlike in the case of SCA, the International Huntington Association (IHA) and the World Federation of Neurology (WFN) jointly issued guidelines for the predictive testing for Huntington disease in 1994.26 These recommendations are successful as the minimum parameters to be considered in patients with HD, in addition to being useful in other neurodegenerative diseases such as frontotemporal dementia and SCA.5 Analysis of the information gathered in our review of these articles, together with useful existing guidelines for other diseases, could lay the groundwork for issuing specific recommendations for PSD in SCA. These recommendations may include making a register of results in order to analyse the benefits of these data at a later time.
ConclusionsPresymptomatic diagnosis of SCA is an opportunity for at-risk subjects to know if they will develop the disease in later life; it also helps them plan their families in light of this risk. Presymptomatic diagnosis should be performed following guidelines intended to safeguard subjects’ physical and psychological health. To date, there are no specific guidelines on performing PSD in patients at risk for SCA. PSD is currently undertaken following guidelines issued by international organisations and additional recommendations designed for other diseases. Implementing genetic counselling programmes is essential in all hospitals aiming to diagnose SCA before symptoms manifest. An ethics committee must approve, regulate, and ensure fulfilment of procedures used in PSD; the participation of the ministry of health and support from specialised organisations, where available, are also crucial. Whenever possible, molecular diagnosis of SCA should be performed exclusively in patients older than 18 with a clinical suspicion of the disease and a minimum risk of 50%, i.e., when one of the parents is affected. Furthermore, a genetic counselling programme including a multidisciplinary team of at least a geneticist, a neurologist, and a psychologist and/or a psychiatrist should be available.
FundingThis article has received no public or private funding.
Conflicts of interestThe authors have no conflicts of interest to declare.
We would like to thank Dr María Cristina Moran Moguel.
Please cite this article as: Orozco-Gutiérrez MH, Cervantes-Aragón I, García-Cruz D. Consideraciones éticas en el diagnóstico presintomático de ataxias espinocerebelosas autosómico dominantes. Neurología. 2017;32:469–475.
This study has not been presented at any of the SEN Annual Meetings or at any other events.
articles

Neurología (English Edition) follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals