




Friedreich ataxia (FA) is the most frequent autosomal recessive ataxia, and the most prevalent of all the hereditary ataxias (2-4 cases per 100000 population).1–3 Symptoms include progressive gait/limb ataxia with motor weakness, areflexia in the lower limbs, loss of proprioception, dysarthria, nystagmus, and auditory alterations. Initial symptoms normally manifest during puberty, although onset may occur from early childhood (2-3 years) to adulthood (> 25 years). FA progresses until the patient loses the ability to walk, 10-15 years after symptom onset.2,4 Cardiac involvement occurs in 90%-100% of patients and is secondary to the infiltrating process; it ranges from minor electrocardiographic (ECG) alterations to arrhythmias and such structural alterations as left ventricular hypertrophy and interstitial fibrosis with significant collagen proliferation and fatty degeneration, a frequent cause of early death in these patients.1,5–7
Most of these cases of FA are caused by a large GAA triplet repeat expansion within the first intron of the frataxin (FXN) gene, located on chromosome 9q21.11.1,8 Frataxin is a mitochondrial protein that activates assembly of iron–sulfur (Fe/S) clusters as part of a multiprotein complex. Although its precise physiological function is yet to be established, it is thought to play an essential role in mitochondrial integrity and functioning, and in cellular iron metabolism.2 Frataxin is also involved, probably indirectly, in the signalling of antioxidant defence systems and pathways controlling cell survival or death.2,4 Frataxin deficiency is associated with alterations to Fe/S cluster biogenesis and iron homeostasis, as well as increased levels of oxidative stress, ultimately leading to neurodegeneration and heart disease.2,4
Despite ongoing research into possible disease-modifying drugs, focusing on compounds increasing frataxin levels or decreasing deficiency (antioxidants, etc.), there is currently no curative treatment for FA.9–12 Therefore, the therapeutic options are limited to prevention and treatment of the neurological complications associated with disease progression (physiotherapy, occupational therapy, orthopaedic treatment of scoliosis, etc.) and non-neurological comorbidities, such as heart disease (use of beta-blockers or angiotensin converting enzyme inhibitors) or diabetes.
Insulin-like growth factor 1 (IGF-1) stimulates growth and cell proliferation and inhibits programmed cell death. IGF-1 has shown neuroprotective effects, as well as benefits in the treatment of several hereditary neurodegenerative diseases caused by CAG trinucleotide repeats (e.g., SCA3, SCA6, and SCA7) and in a proof-of-concept clinical trial of FA, probably due to increased frataxin levels and improved mitochondrial activity.13–18 Although the available data are limited, we decided to start treatment with recombinant human IGF-1 (rhIGF-1) (Increlex®, IGF-1 [rDNA], 10mg/mL vial; Ipsen Pharma SA, Spain) in a child with FA, in the context of a compassionate use programme.
We prospectively assessed the safety and effectiveness of subcutaneous rhIGF-1 treatment (initial dose: 25μg/kg twice daily) in a 13-year-old girl with a diagnosis of FA, treated with antioxidant therapy (coenzyme Q10 with vitamin E, idebenone) since the age of 9, as part of a compassionate use programme. The study was approved by our centre's ethics committee, and the patient and her family members signed informed consent forms.
We prepared a protocol for assessment before starting treatment; this included a complete neurological examination, laboratory tests (haematology, biochemistry, liver and kidney function test, serum IGF-1 levels, etc.), ECG, echocardiography, and chest and abdominal ultrasound. After onset of rhIGF-1 therapy, we scheduled quarterly appointments during the first year of follow-up and further appointments every 6 months, to the end of a 36-month period.19 Effectiveness was assessed using the Scale for the Assessment and Rating of Ataxia (SARA)20; all items were filmed. This footage was subsequently reviewed by an independent reviewer who was blinded to the dates of the videos. Regarding treatment safety, we monitored capillary blood glucose using a glucose metre for the first month of treatment, and then after every dose increase to monitor for possible hypoglycaemia. We also scheduled laboratory tests, serum IGF-1 determination, ECG, echocardiography, and abdominal ultrasound every 6 months.
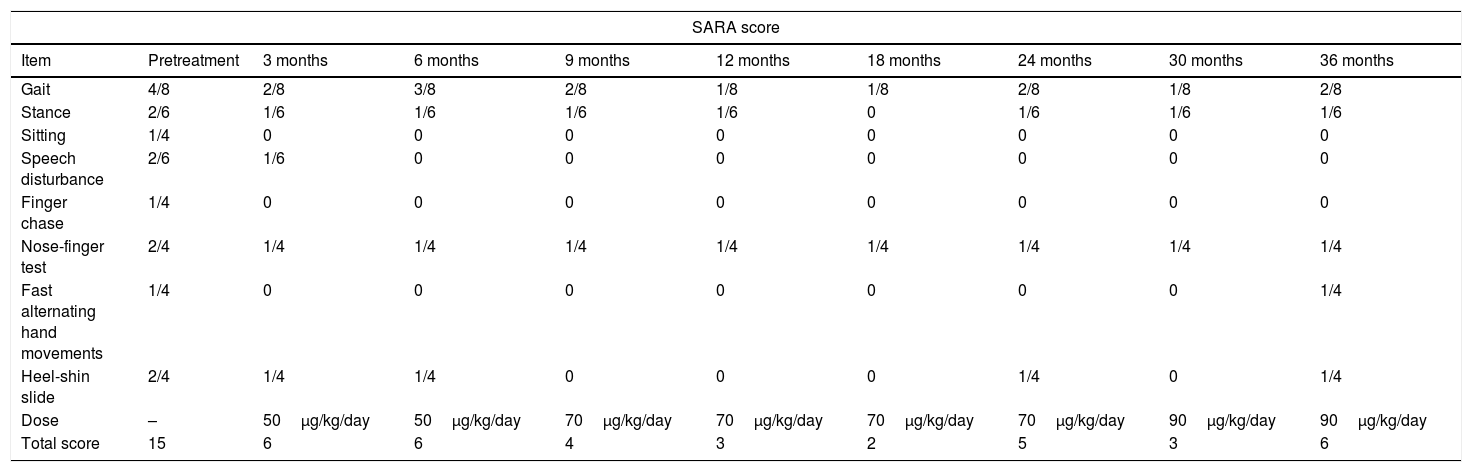
We observed slowing of the progression of neurological impairment during the study period, as well as improved gait (pattern and velocity), balance (fewer falls), and exercise tolerance. The patient began to speak more fluently and performed better in tasks involving fine motor skills, such as handwriting, and increased writing speed. Although the impact on her quality of life was not measured with a formal scale, the treatment had a subjective positive impact on her self-esteem and mood. Symptom improvement was observed from the first months of treatment with rhIGF-1. Total SARA score progressively decreased from the baseline situation (15/40) to the end of the study (6/40), with some fluctuations requiring dose increases. Table 1 shows the total and item scores for the SARA scale, as well as the rhIGF-1 dose. The independent reviewer also observed a clear improvement in motor coordination and balance in the videos, and agreed with the SARA ratings.
Neurological outcomes according to the Scale for the Assessment and Rating of Ataxia (SARA).
| SARA score | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Item | Pretreatment | 3 months | 6 months | 9 months | 12 months | 18 months | 24 months | 30 months | 36 months |
| Gait | 4/8 | 2/8 | 3/8 | 2/8 | 1/8 | 1/8 | 2/8 | 1/8 | 2/8 |
| Stance | 2/6 | 1/6 | 1/6 | 1/6 | 1/6 | 0 | 1/6 | 1/6 | 1/6 |
| Sitting | 1/4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Speech disturbance | 2/6 | 1/6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Finger chase | 1/4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Nose-finger test | 2/4 | 1/4 | 1/4 | 1/4 | 1/4 | 1/4 | 1/4 | 1/4 | 1/4 |
| Fast alternating hand movements | 1/4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1/4 |
| Heel-shin slide | 2/4 | 1/4 | 1/4 | 0 | 0 | 0 | 1/4 | 0 | 1/4 |
| Dose | – | 50μg/kg/day | 50μg/kg/day | 70μg/kg/day | 70μg/kg/day | 70μg/kg/day | 70μg/kg/day | 90μg/kg/day | 90μg/kg/day |
| Total score | 15 | 6 | 6 | 4 | 3 | 2 | 5 | 3 | 6 |
An echocardiography revealed a slight improvement of the septal hypertrophy present at the beginning of the study, which remained stable until the end. The ventricular septum during late diastole decreased from 9mm at baseline to 7.2mm at one year of treatment (Fig. 1A and B), with no systolic or diastolic ventricular dysfunction. Baseline electrocardiography revealed non-specific repolarisation alterations with left precordial T wave inversion and no pathological Q waves (Fig. 1C), a typical sign of FA; this persisted until the end of the study.
 Baseline echocardiography. (B) Echocardiography one year after onset of treatment with rhIGF-1: slight reductions are observed in the ventricular septum and in the diastolic diameter of the left ventricle. (C) Changes in repolarisation manifested as changes in T waves with no pathological Q waves.")
(A) Baseline echocardiography. (B) Echocardiography one year after onset of treatment with rhIGF-1: slight reductions are observed in the ventricular septum and in the diastolic diameter of the left ventricle. (C) Changes in repolarisation manifested as changes in T waves with no pathological Q waves.
Regarding safety, no adverse events related to rhIGF-1 therapy were observed, with the exception of minor inflammatory reactions at the injection site, lasting 4-5 days. The maximum serum IGF-1 concentration with full doses of rhIGF-1 was 1071ng/mL (normal range, 180-1000ng/mL). The patient's glycaemic profile was normal throughout the study.
To our knowledge, this is the first patient with FA treated with rhIGF-1 presenting a symptomatic clinical improvement with subsequent stability at 3 years of follow-up. Treatment was started at onset of neurological impairment; the greatest clinical improvement in neurological status was observed in the first 3 months, with a decrease of 9 points on the SARA scale. It is important to highlight that the initial clinical improvement progressed over the following 12 months and remained stable despite small oscillations in the SARA scores, which were treated by progressively increasing rhIGF-1 doses until reaching a total dose of 90μg/kg/day at 36 months. This neurological improvement permitted total independence for such daily activities as dressing, eating, writing and exercising, until the end of the study.
Although monitoring the patient's heart disease was not a primary objective in our study, echocardiography findings revealed that not only did treatment with rhIGF-1 not increase cardiac hypertrophy, it actually decreased the intraventricular septum during the late diastole. Despite the interesting data on the benefits of this treatment for heart disease in our patient, results should be interpreted with caution, as this is an isolated case. Although the treatment seems to significantly increase frataxin levels in cardiomyocytes, at least in a transgenic mouse model (YG8R mice),21 there is a need for further well-designed studies with larger sample sizes that may confirm its actual usefulness in these patients.
This study supports the hypothesis of the possible neuroprotective effect of treatment with rhIGF-1 on degenerative cerebellar ataxias; this is consistent with data published by a Spanish group in an open-label trial with rhIGF-1 in adults with spinocerebellar ataxia: Arpa et al.16 managed to stabilise disease progression (according to SARA score) for at least the 20-month treatment period.
However, the effectiveness of treatment with rhIGF-1 in patients with FA is not clearly established, and considering that our findings are based on a single clinical case, it should be stressed that they may not be applicable to all cases of FA. Despite this, our data do suggest that treatment with rhIGF-1 may reduce or even improve progressive neurological impairment and halt/improve the associated cardiomyopathy.
As no curative treatment is currently available for FA, we believe that any drug potentially capable of halting or delaying disease progression without causing severe adverse effects merits further research. Further randomised controlled trials are needed to confirm the benefits obtained in our case, to try to determine the action mechanism of rhIGF-1, and to establish what can be expected during the long-term treatment of this and other neurodegenerative diseases.
Please cite this article as: García Ron A, Rodriguez Mesa M. Utilidad del tratamiento como uso compasivo de mecasermina (factor de crecimiento insulínico recombinante humano tipo 1) en ataxia de Friedreich. Neurología. 2020;35:140–143.
