




Behavioural variant frontotemporal dementia (bvFTD) is the most frequent presentation in the clinical spectrum of frontotemporal dementia (FTD) and it is characterised by progressive changes in personality and conduct. Major breakthroughs in molecular biology and genetics made during the last two decades have lent us a better understanding of this syndrome, which may be the first manifestation in many different neurodegenerative diseases.
DevelopmentWe reviewed the main epidemiological, clinical, diagnostic and therapeutic aspects of bvFTD. Most cases manifest sporadically and the average age of onset is 58 years. Current criteria for bvFTD propose three levels of diagnostic certainty: possible, probable, and definite. Clinical diagnosis is based on a detailed medical history provided by family members and caregivers, in conjunction with neuropsychological testing. Treatments which have been used in bvFDT to date are all symptomatic and their effectiveness is debatable. New drugs designed for specific molecular targets that are implicated in frontotemporal lobar degeneration are being developed.
ConclusionsBvFDT is a frequent cause of dementia. It is a non-specific syndrome associated with heterogeneous histopathological and biomolecular findings. The definition of clinical subtypes complemented by biomarker identification may help predict the underlying pathology. This knowledge, along with the development of drugs designed for molecular targets, will offer new treatment possibilities.
La variante conductual de la demencia frontotemporal (DFT vc) es el síndrome clínico más frecuente de las demencias frontotemporales (DFT) y se caracteriza por una alteración progresiva de la personalidad y la conducta. En las últimas 2 décadas, los avances en biología molecular y genética han contribuido a un mayor conocimiento de esta entidad, que puede ser el modo de presentación de diferentes enfermedades neurodegenerativas.
DesarrolloSe revisan los principales aspectos epidemiológicos, clínicos, diagnósticos y terapéuticos de la DFT vc. La mayoría de los casos son esporádicos, iniciándose en torno a los 58 años de media. Los criterios diagnósticos vigentes establecen 3 niveles de certeza diagnóstica: posible, probable y definitivo. El diagnóstico clínico se basa en la anamnesis detallada de familiares, complementada con la realización de test neuropsicológicos dirigidos. Hasta la fecha, los tratamientos empleados son solo sintomáticos y de eficacia controvertida. Se están diseñando fármacos dirigidos contra dianas moleculares específicas implicadas en la patogenia de las degeneraciones lobares frontotemporales.
ConclusionesLa DFT vc es una causa frecuente de demencia. Se trata de un síndrome amplio, heterogéneo desde el punto de vista histopatológico y biomolecular. La definición de subtipos clínicos y la identificación de biomarcadores podrían ayudar a predecir la afección subyacente, lo que junto con el desarrollo de fármacos dirigidos contra dianas moleculares ofrece nuevas posibilidades terapéuticas.
Since Pick described the first case in 1892, it has taken nearly a century for neurology to renew its interest in frontotemporal dementia (FTD).1 In the past 20 years, developments in molecular biology and genetics have produced an undeniable revolution in our knowledge of different types of FTDs. This has enabled researchers to make significant progress in understanding their causal mechanisms. As a result, new diagnostic criteria and classifications that give shape to the current FTD classification scheme have been drafted.
FTDs are listed as the third most common cause of degenerative dementia after Alzheimer disease (AD) and dementia with Lewy bodies. In patients younger than 65, they represent the second most common cause.2,3 Population studies have shown that FTD prevalence rates range between 2.7/100000 (with a peak of 9.4/100000 in subjects aged 60-69) in the Netherlands3 and 15.1/100000 in adults younger than 65 in Cambridge (UK). In this last age group, AD prevalence was the same.2 Although it has traditionally been considered a rare cause of dementia in subjects over 65, it is probably more frequent than previously believed. Some authors identify FTD in 20-25% cases of dementia in patients older than 65 years.2,4,5 Onset normally takes place in the sixth decade of life although this may vary greatly and cases have been reported in patients aged between 30 and 90.2,4,5
Lack of homogeneity in terminology has added to the confusion for years. Frontotemporal dementia is a clinical term that refers to the group of syndromes characterised by progressive decline in behaviour or language and associated with focal atrophy of the frontal and temporal lobes. Predominant symptoms and their time of onset during the course of the disease define 3 main clinical syndromes: behavioural variant of FTD (bvFTD), semantic dementia (SD), and non-fluent primary progressive aphasia (nfPPA).6 Patients in whom FTD is associated with signs of motor neuron disease are diagnosed with FTD/MND.7,8 Furthermore, another 2 syndromes are closely related to FTD: corticobasal syndrome and progressive supranuclear palsy. All 6 clinical syndromes are linked to a heterogeneous group of molecular disorders characterised by cortical neurodegeneration, neuronal loss, and microspongiosis of frontal and temporal lobes. These syndromes are classified as frontotemporal lobar degeneration (FTLD). The term FTD is therefore a clinical concept, while FTLD refers to a pathological concept. In this article, we will review clinical, diagnostic, and therapeutic aspects of bvFTD.
Clinical considerations of behavioural variant frontotemporal dementiaThe most frequent clinical syndrome of FTD is bvFTD. It is characterised by the early onset (within the first 3 years) of insidious changes in personality and behaviour. There are 3 clinical subtypes corresponding to the affected prefrontal areas: dorsolateral (dysexecutive syndrome, pseudodepression, or frontal convexity syndromes), orbitomedial (disinhibition syndrome, pseudomania, or pseudopsychopathy), or medial frontal/cingulate gyrus (apathetic syndrome, akinetic syndrome).9,10 Onset of the disease usually occurs before the age of 65 and typical age of onset is about 58 years.4 Nevertheless, time of onset is frequently hard to determine since these patients present poor insight; detection of early symptoms will therefore depend on the powers of observation of their relatives and caregivers.
As mentioned before, bvFTD is characterised by insidious changes in personality, interpersonal conduct, and emotional modulation. This is the result of progressive disintegration of the neuronal circuits involved in social cognition, emotional self-regulation, motivation and decision-making.11–15 Apathy is a very frequent symptom and manifests as loss of motivation and interest in personal activities, as well as progressive social isolation. Some authors point that apathy is more frequent in late onset bvFTD,16 although there are contradictory results on this topic in the literature.17
Disinhibition is predominant in other cases, which leads patients to commit impulsive acts, such as spending money in excess or behaving in an indiscreet or sexually inappropriate way. Compulsive gambling or, less commonly, hyper-religious ideation have been described as initial manifestations of bvFTD.18,19 Other studies have described patients with sociopathic and deviant behaviours such as violating traffic regulations or assaulting others physically.20,21
Perseveration, with repetitive and stereotyped behaviours (tendency to repeat movements, sentences, stories, or jokes), is another characteristic finding. These patients usually present rigidity and limited mental flexibility, therefore lack of adaptation to new situations or routines is frequent. Affective disorders range from affective flattening and emotional coldness to expansive affect with signs of hypomania. Eating disorders, including hyperphagia, lack of satiety, cravings for sweet food, loss of appetite, and hyporexia, are frequently observed.22 Although the exact mechanisms remain unknown, these changes may be related to hypothalamic dysfunction.23 Psychotic symptoms are very rare and they have been associated with dementia with motor neuron diseases, or early-onset FTLD cases with fused in sarcoma protein (FTLD-FUS), in which they manifest in up to 50%.24–27
Out of all the behavioural manifestations listed above, social disinhibition, euphoria, stereotyped motor behaviours, and changes in eating behaviour are the most useful for distinguishing between bvFTD and other degenerative disorders such as AD.28,29 Therefore, a detailed behavioural evaluation is crucial for diagnosing bvFTD.
In early stages, despite the presence of severe behavioural and personality changes, neuropsychological tests may yield normal results30 since they reflect dorsolateral frontal lobe dysfunction rather than ventromedial dysfunction. For this reason, adding neuropsychological tests to the evaluation could be useful.31
The Mini Mental State Examination is not sufficiently sensitive to identify bvFTD. Addenbrooke's cognitive examination shows changes in 90% of cases at onset.32 The Spanish version of this test has recently been validated. It shows 92% sensitivity for detecting dementia and has been proved useful for discriminating between AD and bvFTD.33,34 It may therefore be considered a good screening tool. These patients are usually oriented, unlike patients with AD.35 Neuropsychological evaluation should include an executive function assessment, as well as evaluation of other cognitive domains that will be useful in differential diagnosis. Examples include memory, language, and visuospatial functions.36 In Spain, these aspects can be evaluated using the different subtests included in the Barcelona Test,37 as well as the different standardised neuropsychological tests within the NEURONORMA project.38,39 Verbal span (digits), Corsi Block-Tapping Test, Trail Making Test,40,41 Verbal Fluency Tests,42,43 Stroop Colour-Word Interference Test, and the Tower of London Test44,45 are some of the main tests used to evaluate the different domains of executive function. Presence of perseverance and confabulation during the testing process is very typical and can help physicians differentiate bvFTD from other disorders.46,47
It was traditionally believed that the early stages of bvFTD were characterised by impaired attention, altered working memory, and executive dysfunction with relatively unaffected language, episodic memory, and visuospatial functions. Altered episodic memory was in fact considered a criterion for exclusion.48 Recent studies have suggested that episodic memory deficits are more frequent than was once thought.49,50 Some studies including histologically confirmed FTLD series have shown that up to 10%-15% of patients present acute memory changes at onset.51,52 This may call into question the requirement, included in the currently applicable criteria, of “relatively intact episodic memory in comparison with executive functions”.
Over the past few years, researchers have described patients who initially present clinical signs of bvFTD but whose condition does not progress to dementia.53 These patients, termed ‘phenocopies’, are generally men and their condition may remain stable, or even improve, over the course of many years.54,55 Absence of executive dysfunction in neurological tests, preserved memory and social cognition, and absence of atrophy (magnetic resonance imaging), or hypometabolism (positron emission tomography) in neuroimaging tests differentiate these cases from true bvFTD.23,54–56
Patients’ recognition of emotions, especially the negative emotions of fear, sadness, or anger, is impaired from early stages.57,58 However, physiological responses to emotional stimuli (such as skin conductance response) may be preserved.59 Difficulty recognising more complex emotions, such as embarrassment, may also be present.60 These changes are not specific to bvFTD and can also be observed in other FTD subtypes, such as SD.
Patients with bvFTD also present changes in different aspects of social cognition. Lack of empathy and emotional coldness are frequent and can be evidenced by specific tests.61 These patients normally present difficulty inferring other people's intentions, understanding other people's points of view, identifying sarcasm, or understanding situations that require moral judgement.62–64
Activities of daily life (ADL) are affected by more severe, earlier-onset changes than in AD or variants in which language is affected,32,65,66 regardless of length of symptoms or results on neuropsychological tests.32,65 Many patients may present changes in ADL from early stages.
Diagnostic criteria for behavioural variant frontotemporal dementiaBehavioural variant FTD may present a diagnostic challenge, especially during its early stages, since many of its symptoms are identical to those found in psychiatric disorders or other types of dementia.25 New diagnostic criteria (Table 1) have been suggested in a recent attempt to improve diagnostic accuracy for bvFTD.67 These more flexible and less restrictive criteria demonstrate higher sensitivity than the previous ones.48,68 Nevertheless, sensitivity seems to decrease in patients aged 65 and older.
Consensus criteria for clinical diagnosis of behavioural variant of FTD (Rascovsky, 2007).67
| Neurodegenerative disease |
| Progressive deterioration of behaviour and/or cognition evidenced by observation or medical history |
| Possible bvFTD |
| At least 3 of the following cognitive or behavioural symptoms are present, persistently or recurrently (A–F): |
| A. Early behavioural disinhibition (3 years) |
| B. Early apathy or inertia (3 years) |
| C. Early loss of empathy (3 years) |
| D. Perseverative, stereotyped, or compulsive/ritualistic behaviour |
| E. Hyperorality and dietary changes |
| F. Neuropsychological profile: executive function deficits with relative sparing of memory and visuospatial functions |
| Probable bvFTD |
| All following conditions should be met: |
| A. Fulfils criteria for possible bvFTD |
| B. Exhibits significant functional decline |
| C. Neuroimaging findings compatible with frontal and/or anterior temporal atrophy (CT or MRI) or hypoperfusion (SPECT) or hypometabolism (PET) |
| Definitive bvFTD |
| Criterion A plus B or C must be met: |
| A. Meets criteria for possible or probable bvFTD |
| B. Histopathological evidence of FTLD in biopsy or at post-mortem |
| C. Presence of a known pathogenic mutation |
| Exclusion Criteria for bvFTD |
| A. Pattern of deficits is better accounted for by a different non-degenerative neurological or other medical disorder |
| B. Behavioural disturbance is better accounted for by a psychiatric disorder |
| C. Biomarkers strongly indicative of Alzheimer disease or another neurodegenerative processa |
bvFTD: behavioural variant frontotemporal dementia; FTLD: frontotemporal lobar degeneration; PET: positron emission tomography; MRI: magnetic resonance imaging; SPECT: single photon emission computed tomography; CT: computed tomography.
Median survival of FTD patients has been estimated at 6-11 years from symptom onset, and 3-4 years from diagnosis.69–71 Most studies suggest that survival rates are lower and cognitive and functional impairment progresses faster in DFT than in AD.70,71 However, other studies suggest the opposite.72 Association with MND (survival of 2.4-4.9 years from onset and 1.2-1.4 years from diagnosis)71 and presence of language impairment at diagnosis have been proposed as factors reducing survival in bvFTD.73
Although it has been used in most studies, the clinical dementia rating (CDR) score,74 was initially designed for AD and mainly assesses memory alteration. It tends to underestimate dementia severity in bvFTD.75 An adapted version including language and behaviour domains (fCDR) is sensitive enough to detect changes in most FTD patients.75 A recently-developed FTD-specific scale (FRS) also includes behavioural and ADL changes.76 According to this scale, bvFTD patients tend to present more severity and faster progression than patients with semantic dementia, regardless of length of symptoms.76
Josephs et al.77 studied 86 patients with bvFTD and analysed possible predictor variables for disease progression. They found that anatomical subtype was the most powerful predictor variable for the course of the disease. Thus, patients who presented faster progression according to the CDR-SB scale showed a predominantly frontal and frontotemporal pattern of atrophy with less pronounced temporal pole volume loss. Other predictors of rapid progression were older age at onset; poorer executive, language and visuospatial function; less disinhibition, agitation/aggression, and night-time behaviours at onset; and mutations in the microtubule associated protein tau (MAPT) gene.77
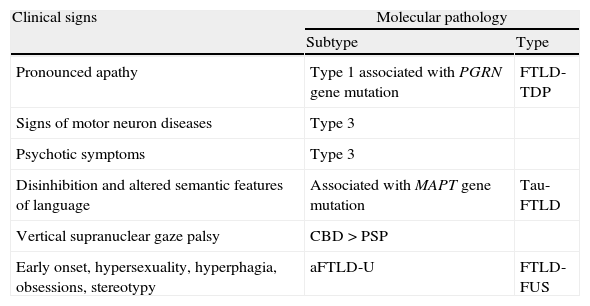
More than clinical symptomsClinical symptoms are essential, but not sufficient, to diagnose bvFTD. First, assigning an early diagnosis of bvFTD may be difficult: changes in patient's social cognition may go unnoticed until they interfere with the caregiver's duties. Also, the patient's cognitive performance may be normal during the first stages, and neuropsychiatric symptoms can sometimes be erroneously attributed to a primary psychiatric disorder. As we have already mentioned, there are also patients with symptoms compatible with bvFTD whose condition does not finally progress to dementia (phenocopies), and others who fulfil clinical criteria for bvFTD but whose autopsies reveal diseases other than FTLD.78 Second, the new drugs being developed to treat these diseases are agents aimed at specific molecular targets, and therefore in vivo assessment of disease pathology and molecular biology is especially important. Some clinical phenotypes show a strong association with specific histopathological subtypes, but in the case of bvFTD we found no clear correlations with any particular subtypes.78,79 Therefore, while some clinical signs suggesting specific molecular subtypes have been identified (Table 2),79 it is also possible that a correlation between different subtypes and certain anatomical and biochemical features may exist.78
Clinical signs indicative of certain biomolecular subtypes in bvFTD.
| Clinical signs | Molecular pathology | |
| Subtype | Type | |
| Pronounced apathy | Type 1 associated with PGRN gene mutation | FTLD-TDP |
| Signs of motor neuron diseases | Type 3 | |
| Psychotic symptoms | Type 3 | |
| Disinhibition and altered semantic features of language | Associated with MAPT gene mutation | Tau-FTLD |
| Vertical supranuclear gaze palsy | CBD>PSP | |
| Early onset, hypersexuality, hyperphagia, obsessions, stereotypy | aFTLD-U | FTLD-FUS |
FTLD-TDP subtypes 1 and 3 have been designated according to Mackenzie classification.
Taken from data by Josephs et al. (2007).79
aFTLD-U: atypical frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions; CBD: corticobasal inclusion; FTLD-FUS: frontotemporal lobar degeneration associated with fused in sarcoma protein; FTLD-Tau: frontotemporal lobar degeneration with tau inclusions; FTLD-TDP: frontotemporal lobar degeneration associated with TAR DNA-binding protein 43; MAPT: microtubule-associated protein tau; PGRN: progranulin; PSP: progressive supranuclear palsy.
No specific treatment approaches for FTD are currently available in clinical practice. Treatment is basically symptomatic or supportive and its main objective is to alleviate symptoms and improve the patient's quality of life, especially when behavioural changes are so pronounced that they interfere with patient care.80 Since no effective treatments are available, physicians often use psychoactive drugs off-label to alleviate symptoms.81,82 The most commonly used drugs are selective serotonin reuptake inhibitors (SSRI) and antipsychotics, which indicates that serotonergic and dopaminergic changes are associated with FTD.83–85
SSRIs have been used to treat behavioural symptoms, impulsiveness-disinhibition, and eating disorders in patients with FTD, with mixed results.86–89 Despite the lack of scientific evidence, atypical antipsychotics have also been used to treat FTD symptoms, especially agitation and disinhibition. The literature describes one case treated with risperidone90 and an open trial with olanzapine that favours use of the drug.91 Other dopaminergic agents, such as selegiline (IMAO-B),92 have been used successfully to reduce neuropsychiatric symptoms. Methylphenidate has also been tested in patients with bvFTD with satisfactory results.93
Cholinesterase inhibitors and memantine are also frequently used for FTD94,95 because of their effect in AD and based on data from open uncontrolled clinical trials with low patient numbers. However, the poor results obtained with donepezil, together with the controversial results for galantamine and rivastigmine,96–100 have given rise to a general recommendation that cholinesterase inhibitors should be avoided as treatment for FTD.80–82 Identification of 3 bvFTD cases101 that showed an improved score on the neuropsychiatric inventory (NPI) after treatment with memantine encouraged researchers to conduct clinical trials. Two open uncontrolled clinical trials with memantine have been completed. One included a series of 16 patients with bvFTD102 whose behavioural symptoms did not improve with memantine dosed at 20mg/kg/day. In the other study, the same dose of memantine was administered to 21 patients with bvFTD, 13 patients with SD, and 9 with nfPPA.103 Patients with bvFTD demonstrated a temporary initial improvement on the NPI score, but subsequent follow-up did not show any other benefits. Two multicentre, prospective and randomised double-blind placebo-controlled trials are currently being performed (a phase VI trial in the USA and a phase II trial in France).
Disease-modifying treatmentThe most promising line of action focuses on developing drugs that bind to a specific molecular target. In the case of FTLD, the main therapeutic targets are Tau protein, TAR DNA-binding protein 43 (TDP-43), and to a lesser extent, FUS protein. Tau protein participates in the pathogenesis of AD, as well as in a large percentage of all FTLD cases. Therefore, these new Tau aggregation inhibitors being developed for AD may be also useful for treating Tau-FTLD.80,82,104
Progranulin (PGRN) mutations cause haploinsufficiency due to loss of protein function and they are associated with some cases of FTLD, mainly FTLD-TDP.105 Plasma PGRN levels are low in these cases,105–107 so normalising these levels could constitute an interesting therapeutic strategy. Researchers have recently managed to use micro-ARN-29b to increase PGRN levels in vitro.108 Other possible therapeutic targets include inhibition of hyperphosphorylation, ubiquitination, cleavage, and TDP-43 translocation from the cytoplasm to the nucleus.109
ConclusionsFTLDs are frequent causes of dementia. Over the past 20 years, knowledge of this group of diseases has increased considerably thanks to advances in molecular biology and genetics. Behavioural variant FTD, characterised by insidious changes in personality and behaviour, is the most frequent clinical syndrome in this group. This devastating condition typically manifests in middle-aged subjects and affects individuals with active family, social, and professional lives. Assigning an early clinical diagnosis is often difficult and since the patient may show poor insight, his or her medical history as described by relatives is a crucial resource. From a histopathological and biomolecular point of view, bvFTD is a heterogeneous condition and predicting the underlying pathology in vivo is still difficult. The definition of the syndrome describes too broad of a concept. Therefore, identifying differential characteristics that help define ‘clinical subtypes’ may be useful for improving the clinical-pathological correlation. The identification of biomarkers and development of drugs aimed at specific molecular targets are essential strategies that will usher in new treatment approaches.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Fernández-Matarrubia M, Matías-Guiu JA, Moreno-Ramos T, Matías-Guiu J. Demencia frontotemporal variante conductual: aproximación clínica y terapéutica. Neurología. 2014;29:464‐472.
