




Lobar frontotemporal degeneration (FTLD) encompasses a group of molecular disease defined by the deposition of an abnormal protein in the central nervous system. Behavioural variant frontotemporal dementia (bvFTD) is the most frequent clinical presentation of FTLD. The past two decades of research have contributed to a better understanding of this entity, which may be the first manifestation in many different neurodegenerative disorders.
DevelopmentWe reviewed correlations between clinical, pathological, and genetic findings and the main disease biomarkers of FTLD, with particular interest in bvFTD. Anatomical pathology findings in FTLD are heterogeneous and the syndrome is not associated with any one specific histopathological type. Promising available biomarkers include structural and functional neuroimaging techniques and biochemical and genetic biomarkers. Disease-modifying drugs designed for specific molecular targets that are implicated in FTLD pathogenesis are being developed.
ConclusionsBvFTD is a frequent cause of dementia. Of all the clinical variants of FTLD, behavioural variant is the one in which establishing a correlation between clinical and pathological signs is the most problematic. A biomarker evaluation may help predict the underlying pathology; this approach, in conjunction with the development of disease-modifying drugs, offers new therapeutic possibilities.
Las degeneraciones lobares frontotemporales (DLFT) son un grupo de patologías moleculares que se definen en función de la proteína acumulada en el sistema nervioso central. La demencia frontotemporal variante conductual (DFT vc) es el síndrome clínico de presentación más frecuente. Los avances realizados en los últimos años han contribuido a un mayor conocimiento de esta entidad, que puede ser el modo de presentación de diferentes enfermedades neurodegenerativas.
DesarrolloSe revisa la correlación entre clínica, patología y genética de las DLFT, en especial de la DFT vc, así como los principales biomarcadores de la enfermedad. La anatomía patológica de la DFT vc es muy variada, sin mostrar asociación significativa con ningún subtipo histopatológico concreto. Entre los biomarcadores disponibles, destacan la neuroimagen anatómica y funcional, los biomarcadores analíticos y la genética. Se están diseñando fármacos dirigidos contra dianas moleculares concretas implicadas en la patogenia de las DLFT.
ConclusionesLa DFT vc es una causa frecuente de demencia. De entre todas las variantes clínicas de las DLFT, es en la que resulta más difícil establecer una relación clínico-patológica. El uso de biomarcadores puede ayudar a predecir la anatomía patológica subyacente, lo que junto al desarrollo de fármacos ligando-específicos ofrece nuevas posibilidades terapéuticas.
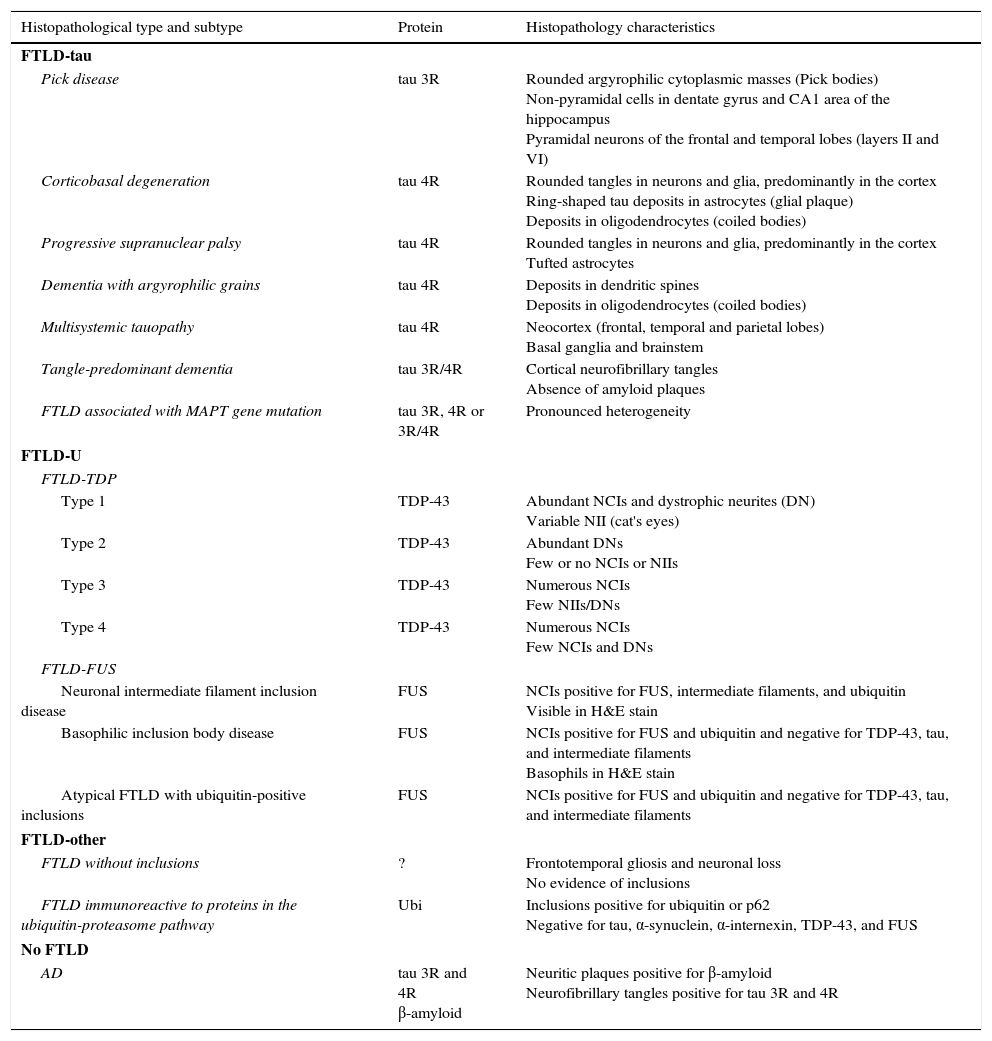
The term frontotemporal lobar degeneration (FTLD) designates a pathological concept that includes a group of molecular diseases that are classified according to the protein that accumulates in the central nervous system. In 2007, Cairns et al.1 established 4 main subtypes according to the protein deposits: tauopathies (FTLD-tau), ubiquitinopathies (FTLD-U), dementia without specific histopathological changes, and neuronal intermediate filament inclusion disease. Since then, following new breakthroughs in pathology and molecular biology, this classification system has been modified. FTLD cases are now classified into 3 main subtypes according to pathology: FTLD associated with tau protein (FTLD-tau), FTLD associated with the TAR DNA-binding protein 43 (FTLD-TDP), and FTLD associated with the fused-in sarcoma protein (FTLD-FUS). Only a very small percentage of cases will not fit any of these subtypes (FTLD-other).2 Each of these main pathological groups can be subdivided into different entities or variants according to the distribution and morphology of protein inclusions and the histological characteristics specific to the case (Table 1). These groups give rise to 6 well-defined syndromes: the 3 clinical variants of frontotemporal dementia or FTD (behavioural variant frontotemporal dementia [bv-FTD], progressive nonfluent aphasia [PNFA], semantic dementia [SD]); frontotemporal dementia associated with motor neuron disease (FTD-MND); progressive supranuclear palsy syndrome (PSPS); and corticobasal syndrome (CBS).
Histopathological and biomolecular classification of types of frontotemporal lobar degeneration
| Histopathological type and subtype | Protein | Histopathology characteristics |
|---|---|---|
| FTLD-tau | ||
| Pick disease | tau 3R | Rounded argyrophilic cytoplasmic masses (Pick bodies) Non-pyramidal cells in dentate gyrus and CA1 area of the hippocampus Pyramidal neurons of the frontal and temporal lobes (layers II and VI) |
| Corticobasal degeneration | tau 4R | Rounded tangles in neurons and glia, predominantly in the cortex Ring-shaped tau deposits in astrocytes (glial plaque) Deposits in oligodendrocytes (coiled bodies) |
| Progressive supranuclear palsy | tau 4R | Rounded tangles in neurons and glia, predominantly in the cortex Tufted astrocytes |
| Dementia with argyrophilic grains | tau 4R | Deposits in dendritic spines Deposits in oligodendrocytes (coiled bodies) |
| Multisystemic tauopathy | tau 4R | Neocortex (frontal, temporal and parietal lobes) Basal ganglia and brainstem |
| Tangle-predominant dementia | tau 3R/4R | Cortical neurofibrillary tangles Absence of amyloid plaques |
| FTLD associated with MAPT gene mutation | tau 3R, 4R or 3R/4R | Pronounced heterogeneity |
| FTLD-U | ||
| FTLD-TDP | ||
| Type 1 | TDP-43 | Abundant NCIs and dystrophic neurites (DN) Variable NII (cat's eyes) |
| Type 2 | TDP-43 | Abundant DNs Few or no NCIs or NIIs |
| Type 3 | TDP-43 | Numerous NCIs Few NIIs/DNs |
| Type 4 | TDP-43 | Numerous NCIs Few NCIs and DNs |
| FTLD-FUS | ||
| Neuronal intermediate filament inclusion disease | FUS | NCIs positive for FUS, intermediate filaments, and ubiquitin Visible in H&E stain |
| Basophilic inclusion body disease | FUS | NCIs positive for FUS and ubiquitin and negative for TDP-43, tau, and intermediate filaments Basophils in H&E stain |
| Atypical FTLD with ubiquitin-positive inclusions | FUS | NCIs positive for FUS and ubiquitin and negative for TDP-43, tau, and intermediate filaments |
| FTLD-other | ||
| FTLD without inclusions | ? | Frontotemporal gliosis and neuronal loss No evidence of inclusions |
| FTLD immunoreactive to proteins in the ubiquitin-proteasome pathway | Ubi | Inclusions positive for ubiquitin or p62 Negative for tau, α-synuclein, α-internexin, TDP-43, and FUS |
| No FTLD | ||
| AD | tau 3R and 4R β-amyloid | Neuritic plaques positive for β-amyloid Neurofibrillary tangles positive for tau 3R and 4R |
FTLD-TDP subtypes 1-4 are designated according to Mackenzie's classification.
AD: Alzheimer disease; FTLD-FUS: frontotemporal lobar degeneration associated with fused-in sarcoma protein; FTLD-tau: frontotemporal lobar degeneration with tau inclusions; FTLD-TDP: frontotemporal lobar degeneration associated with TAR DNA-binding protein 43; aFTLD-U: atypical frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions; NCI: neuronal cytoplasmic inclusions; NII: neuronal intranuclear inclusions; Ubi: ubiquitin.
A small percentage of cases with clinical features suggesting FTD actually have Alzheimer disease (AD).3 This finding is more frequent in cases of logopenic primary progressive aphasia (PPA),4 but it has also been described in some cases of SD (10%)5 and bv-FTD (5%-7%).5–7 Cases of AD with an antemortem diagnosis of bv-FTD are referred to as frontal-variant AD.8
The purpose of this study is to revise the clinical-pathological and biomolecular correlations in FTD subtypes, especially bv-FTD, as well as the diagnostic accuracy of biomarkers used to hypothesise which disease was responsible for clinical symptoms.
Clinical-pathological and biomolecular correlations in frontotemporal lobar degenerationFTLDs have classically been regarded as a heterogeneous group of neurodegenerative diseases and researchers have found it extraordinarily difficult to establish links between clinical presentation and the underlying pathological process. Doctors also believed it was not possible to deduce the type of clinical manifestation based on the pathological diagnosis. Advances in neuropathology, biochemistry, molecular biology, and genetics now let us establish clinical, histological, biomolecular, and genetic correlations, in addition to links between clinical and pathological findings.
In 2011, Josephs et al.9 published a review of several clinical-pathological studies10–17 performed in the preceding decade in 6 hospitals in the United States, Canada, the UK, and Australia, including a total of 544 cases of FTLD. They observed that bv-FTD showed similar percentages of underlying FTLD-tau and FTLD-TDP. Almost all cases of PSPS and CBS presented FTLD-tau, whereas 100% of the cases of FTD-MND displayed FTLD-TDP. FTLD-TDP was present in 83% of the patients with SD, whereas PNFA was fundamentally associated with FTLD-tau (70%). Examining by specific groups (FTLD-TDP and FTLD-tau), these researchers found significant associations between clinical phenotype and histopathological subtype, except for PNFA and the FTLD-tau subtype, for which associations were not statistically significant. Patients with bv-FTD and tauopathy were associated with Pick disease (PiD) in 70% of all cases. Those with TDP histopathology demonstrated an association with subtype 1 in the Mackenzie et al. classification.18 Nevertheless, the association between bv-FTD and the histopathological subtype was not robust (Fig. 1).

Clinical, molecular, and genetic correlations in frontotemporal lobar degeneration. Arrows indicate the links between clinical syndromes and underlying disease, while continuous lines show the most robust associations. The most frequent histopathological subtype is listed for each syndrome. FTD: frontotemporal dementia; FTLD: frontotemporal lobar degeneration; PSPS: progressive supranuclear palsy syndrome; CBS: corticobasal syndrome; PNFA: progressive nonfluent aphasia; bv-FTD: behavioural variant frontotemporal dementia; SD: semantic dementia; FTD-MND: frontotemporal dementia associated with motor neuron disease; PSP: progressive supranuclear palsy; CBD: corticobasal degeneration; PiD: Pick disease; MAPT: microtubule associated protein tau; FUS: fused-in sarcoma protein; PGRN: progranulin; VCP: valosin containing protein; TARDBP: TAR DNA-binding protein 43; CHMP2B: charged multivesicular body protein 2B. FTLD-TDP subtypes 1-3 are designated according to Mackenzie's classification.
In some cases of bv-FTD, doctors may find clinical features that point towards a specific histopathological subtype.19 One clinical subtype is characterised by changes in behaviour and personality, especially hypersexuality, hyperphagia, and stereotyped or obsessive behaviour. This subtype is strongly associated with atypical FTLD with immunoreactivity to ubiquitin only (aFTLD-U).20,21 These cases also show very early onset (mean age of about 40 years), and they are typified by pronounced striatal atrophy.22,23 On the other hand, the coexistence of psychotic symptoms or signs of motor neuron disease (MND) points to FTLD-TDP type 3 pathology,24 whereas apathy as the dominant trait is indicative of FTLD-TDP type 1 associated with PGRN mutations.25,26 The coexistence of marked disinhibition and alteration of the semantic axis of language suggests tau pathology associated with mutation of the microtubule-associated protein tau (MAPT) gene.14,26,27 The presence of vertical supranuclear gaze palsy is associated with tau pathology, and specifically with corticobasal degeneration; to a lesser extent, it is associated with progressive supranuclear palsy (PSP).28
Although the clinical-pathological correlations for the FTLD-FUS subtype have undergone less study,29–36 it seems there is also an association between clinical phenotype and FTLD-FUS. Neuronal intermediate filament inclusion disease has been linked to 3 syndromes; the most frequent is bv-FTD, followed by FTD-MND and CBS/primary lateral sclerosis. Basophilic inclusion body disease is associated with bv-FTD, FTD-MND, motor neuron diseases, and juvenile amyotrophic lateral sclerosis (JALS). There is a strong association between aFTLD-U and bv-FTD. There are no clinical-pathological studies on FTLD without inclusions, or on FTLD with cytoplasmic inclusions that are immunoreactive to proteins in the ubiquitin-proteasome pathway and negative for TDP-43 (FTLD-UPS).
Although one specific syndrome tends to be dominant in the first 4 years, it is common for different clinical syndromes in FTD to overlap as the disease progresses.37 Kertesz et al.38 prospectively studied 262 patients with clinical criteria of FTD. During follow-up, they documented the appearance of second and sometimes even third clinical syndromes. Patients with bv-FTD developed PNFA in 50% of the cases, CBS/PSPS in 22.5% of the cases, and SD in about 16% of the cases. Slightly more than 50% of the PNFA cases developed bv-FTD and a third developed CBS/PSPS. The rest of the PNFA cases did not exhibit any other syndromes. Three-quarters of the patients with SD developed bv-FTD and the rest did not progress to exhibit other clinical syndromes. Cases with CBS/PSPS were likely to develop bv-FTD (50%) or PNFA (50%).
Since the same patient may manifest different clinical syndromes as the disease progresses, it is possible to state that the same histopathological subtype may elicit different types of clinical disease due to unknown aetiological or neuropathological mechanisms.
Clinical-pathological correlations in the genetic variants of frontotemporal lobar degenerationAlthough most FTLD cases are sporadic, a small percentage of them are associated with specific genetic mutations. Seven genes are currently known to be involved in cases of familial FTLD: MAPT, the progranulin gene (PGRN), C9ORF72, valosin containing protein-1 (VCP-1), charged multivesicular body protein 2B (CHMP2B), TAR DNA-binding protein 43 (TARDBP) and fused-in sarcoma (FUS). Mutations in the MAPT, PGRN, and C9ORF72 are responsible for most FTD cases with an autosomal dominant inheritance pattern.
MAPT mutations39 are typically associated with FTLD-tau pathology and do not display a specific histological pattern;40 histological presentations may include PiD, PSP, and corticobasal degeneration (CBD).41 Patients with MAPT gene mutations usually present a bv-FTD phenotype, which is occasionally associated with extrapyramidal symptoms.27 This group characteristically presents pronounced symmetrical atrophy of the anterior medial temporal and orbitofrontal regions,42,43 and it is therefore quite common for the disease to manifest as SD with signs of bv-FTD (or bv-FTD with changes in the semantic processing of language). Disinhibition is a particularly common feature among these patients.23,26,27
Familial cases associated with PGRN mutations present a typical neuropathology pattern with marked atrophy at the level of the frontal lobe, caudate nucleus, substantia nigra, and medial area of the thalamus. Unlike MAPT mutations, which are associated with tauopathy, PGRN mutations are associated with deposits of TDP-43, especially in type 1 FTLD-TDP according to Mackenzie's classification.18 Neuronal intranuclear inclusions with a ‘cat's eye’ or ‘lentiform’ appearance are the most striking histological finding.44–46
Clinically, expression of PGRN mutations varies considerably, even within the same family in addition to between different families. Age at onset is also quite variable, once again, even among members of the same family.47 These mutations may present as CBS,48–51 PNFA,23,51,52 and bv-FTD,48 with apathy as the dominant trait.25 Cases of associated MND are exceptional.53 In patients with PGRN mutations, even when asymptomatic, plasma levels of PGRN are very low.47,54–56
Hexanucleotide repeat expansion of the nucleotide string GGGGCC in the first intron of the C9ORF72 gene is not only the known mutation most frequently associated with ALS and FTD-MND, but also the second-most frequent in FTLD after PGRN mutations. According to DeJesus-Hernandez et al.,57 12% of the familial cases of FTD and 22.5% of ALS cases are associated with that mutation. Slightly higher prevalences have been found in Nordic populations: the mutation was present in 46% of the population with familial ALS, 21.1% in sporadic ALS, and 29.3% in FTD in Finland.58
Mutations in the C9ORF72 gene, associated with FTLD-TDP pathology, may manifest clinically as bv-FTD, ALS, or FTD-MND. Presentations vary considerably, even between members of the same family. In the study by DeJesus-Hernandez et al.,57 26.9% of the patients with FTLD also presented ALS, and more than 30% had family members with ALS.
Mutations in the VCP-1 gene are associated with a rare autosomal dominant disorder that presents with myopathy (90%), Paget's disease of bone, and less frequently, FTD (30%). FTD usually appears years after the onset of muscular symptoms.59 According to Sampathu's and Mackenzie's histopathological classification systems for types of FTLD-TDP, the above entity is known as subtype 4.60 Biopsy typically reveals myopathic inclusion bodies.
Mutations in the CHMP2B gene, which until now have only been detected in 5 families, are typically associated with bv-FTD, with or without associated pyramidal or extrapyramidal signs.61,62 Myoclonia may also appear in later stages.61 There are only 2 published cases of associated MND.63 From a neuropathology viewpoint, they are associated with FTLD-UPS.
Mutations in the TARDBP and FUS genes are fundamentally associated with cases of familial ALS and only rarely do they cause FTD.64–67
The relationship between different clinical phenotypes and underlying disease in genetic cases differs from that found in sporadic cases. For example, PNFA and CBS in sporadic cases are more commonly associated with FTLD-tau, whereas in familial cases, the same symptoms would be more frequently found in FTLD-TDP (especially type 1). Either sporadic or familial bv-FTD may be associated with FTLD-tau or FTLD-TDP indiscriminately. Cases of SD and PSPS are almost always sporadic.
Neuroimaging biomarkersStructural neuroimaging: brain magnetic resonanceNeurodegenerative diseases are associated with progressive cerebral atrophy. This process can be measured and quantified using structural neuroimaging techniques, including brain MRI. Measuring atrophy using neuroimaging techniques has been shown to be a useful biomarker in other neurodegenerative diseases.68,69
The relationship between different FTD syndromes and the pattern of atrophy is now well-defined. In general, bv-FTD is associated with atrophy predominantly affecting the frontal lobe, insula, anterior cingulate, and anterior temporal lobe in an asymmetrical pattern (predominantly in the non-dominant hemisphere). SD is associated with asymmetrical atrophy affecting the anterior inferior temporal lobes, whereas PNFA is associated with asymmetrical atrophy of the anterior perisylvian cortex (more pronounced in the dominant hemisphere).70–72
Lindberg et al.73 analysed the volumes of 9 cortical regions of interest in 61 individuals (27 controls, 12 with bv-FTD, 9 with PNFA, and 13 with SD) and compared findings between the different clinical entities. They observed that it was possible to distinguish between these syndromes with relatively high levels of sensitivity and specificity. Identifying atrophy as temporal or frontal was the best discriminator between SD and the other 2 clinical subtypes. Laterality of atrophy (right vs left) was the most useful feature for discriminating between bv-FTD and PNFA. Despite the above, doctors must be mindful that there is a certain degree of heterogeneity and overlap between neuroanatomical findings, and that brain MRI may not show alterations in early stages of disease.
Cerebral alterations found in bv-FTD are not limited to the cerebral cortex. Atrophy also affects numerous subcortical regions, including the amygdala, hippocampus, caudate nucleus, striatum, putamen, thalamus, and hypothalamus.74,75 In fact, amygdalar atrophy may be a useful marker for distinguishing between bv-FTD and Alzheimer disease.74 Similarly, Chao et al.76 observed that reduced volume of frontal white matter is indicative of reduction in volume in adjacent grey matter in patients with bv-FTD, and that different FTD subtypes are associated with different white matter atrophy patterns.
Early changes, progression, and stages of behavioural variant frontotemporal dementiaDeveloping automated quantitative methods, such as voxel-based morphometry and cortical thickness mapping, has been crucial for the detection of selective atrophy of the anterior cingulate and the frontal and insular cortices in early stages of bv-FTD.71,77,78 This differs from the atrophy pattern observed in other FTD variants and other types of dementia, including AD.79 Macroscopic changes may not be apparent in early clinical phases, even though molecular disease is known to be present in the brain several years before symptoms appear (preclinical phase). The yearly rate of overall cerebral atrophy in bv-FTD is as high as 8%, or nearly twice that observed in AD. However, some patients with bv-FTD may present lower rates (0.3% yearly) that resemble those seen in healthy individuals.80 This low figure may be due to phenocopies being included among cases.
The advance of brain atrophy in FTD has been studied by analysing the pattern of atrophy in patients with different progression times.81 In initial phases, atrophy generally affects the medial and orbital frontal lobe. It subsequently affects the temporal pole, hippocampal formation, frontal dorsolateral cortex, and basal ganglia. This progression pattern has been linked to the volume of the cortical and subcortical regions, and to underlying neuronal loss.82,83 As a result, determining the atrophy pattern is a useful way of identifying the stage of the disease.84,85 Based on these findings, Kipps et al.86 proposed a visual assessment scale using brain MRI.
Seeley et al.77 carried out a case-control study in patients with bv-FTD at different stages of progression. These researchers classified cases in 3 groups according to scores on the Clinical Dementia Rating scale: CDR 0.5, CDR 1, and CDR 2-3. The group in the mildest stage (CDR 0.5) displayed bilateral but asymmetrical atrophy, predominantly in the right hemisphere, affecting different regions of the frontal lobe (rostromedial, frontal pole, dorsolateral, and orbitofrontal), anterior cingulate cortex, anterior insula, hippocampus, and subcortical areas (ventral striatum, dorsomedial thalamus). Atrophy was more widespread in the above areas in subjects with higher CDR scores, especially in the frontal lobe, and it extended to more posterior areas including the posterior insula and the temporal and parietal lobes. Another study of cases of bv-FTD confirmed by a pathology study showed similar results.87
Subsequent studies suggest that the areas affected in early stages of the disease (frontal, insular, and anterior cingulate) form part of a structurally and functionally differentiated circuit called the salience network (SN). The anatomical substrate of this neural circuit are the von Economo cells, a unique neuronal population that probably plays a fundamental role in social cognition.88,89 A study using functional cerebral MRI found the clinical severity of bv-FTD to be correlated with the degree of disruption in right-sided SN connectivity.90 Nevertheless, given the heterogeneity of bv-FTD pathology, it is not clear whether or not this neural circuit undergoes similar changes in all histopathological subtypes.
Correlation between anatomy and pathological subtypeAn even more interesting prospect is neuroimaging, which has great potential for exploring the pathological process underlying the syndrome.
The first studies carried out provided contradictory results; some showed differences between patients with FTLD-tau and FTLD-U,91 while others found very similar patterns of atrophy.92 In 2009, Whitwell et al.93 used morphometry to analyse regional cortical atrophy in a cohort of 66 patients with a clinical diagnosis of bv-FTD. They observed that half of the patients presented predominantly frontal atrophy, while the other half showed temporal predominance. Based on these findings, they established 4 anatomical subtypes: frontal-dominant, frontotemporal, temporofrontoparietal, and temporal-dominant. The patients with frontal-dominant and frontotemporal atrophy patterns predominantly presented frontal lobe impairment (orbitofrontal, medial frontal, and dorsolateral regions); atrophy was quite restricted in the first pattern, but less restricted and also appearing in temporal areas in the second. Patients with the temporofrontoparietal pattern characteristically showed temporoparietal and medial frontal atrophy. The least frequent subtype was temporal-dominant, in which atrophy was restricted to the inferior and medial region of the temporal lobe. This pattern was associated with predominantly right-sided atrophy in all cases, and with MAPT mutations in 83% of the cases. PGRN mutations, unlike those previously listed, were never associated with the temporal-dominant subtype; this indicates that the anatomical pattern may be useful in distinguishing between MAPT and PGRN mutations. Cases of the temporal-dominant subtype showed poorer results on language and memory tests, and those with the frontal-dominant subtype scored lower for executive function, but differences were not statistically significant. The most frequent behavioural symptom in subtypes with frontal atrophy was apathy (76%), and this is not the case in the temporal-dominant subtype. Among patients who had undergone pathology studies, there were no clear correlations between the anatomical pattern and pathological subtype except in patients diagnosed with FTLD-TDP associated with MND, whose atrophy patterns were restricted to the frontal lobe. In addition to being associated with TDP-43, the temporofrontoparietal pattern was associated with AD or CBD in 3 subjects. According to these authors, the frontotemporal subtype may be a stage in bv-FTD progression; it would begin as frontal-dominant atrophy and subsequently extend to the temporal lobes. The temporofrontoparietal pattern cannot be explained in the same way. Frontal atrophy detected in this pattern was less pronounced than in cases with a frontal-dominant pattern, and temporal atrophy was also less pronounced than in temporal-dominant cases.93
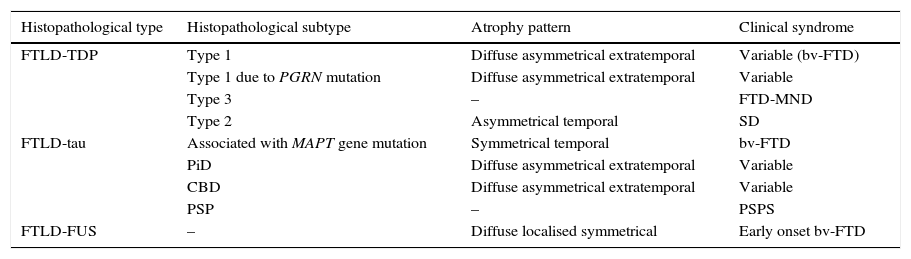
In 2011, Rohrer et al.94 presented a retrospective study in which they analysed clinical, neuropsychological, and neuroimaging data (using voxel-based volumetry and morphometry) in a cohort of 95 patients with a diagnosis of FTLD confirmed by pathology study. These researchers attempted to find clinical-anatomical correlations for all histopathological subtypes. They classified anatomical findings in 4 patterns based on symmetry or asymmetry and the temporal or extratemporal location of atrophy. They observed that specific pathological subtypes exhibited a significant association with distinct clinical syndromes, also finding an association between some subtypes and certain atrophy patterns (Table 2).
Correlations between histopathology, atrophy pattern, and clinical syndrome
| Histopathological type | Histopathological subtype | Atrophy pattern | Clinical syndrome |
|---|---|---|---|
| FTLD-TDP | Type 1 | Diffuse asymmetrical extratemporal | Variable (bv-FTD) |
| Type 1 due to PGRN mutation | Diffuse asymmetrical extratemporal | Variable | |
| Type 3 | – | FTD-MND | |
| Type 2 | Asymmetrical temporal | SD | |
| FTLD-tau | Associated with MAPT gene mutation | Symmetrical temporal | bv-FTD |
| PiD | Diffuse asymmetrical extratemporal | Variable | |
| CBD | Diffuse asymmetrical extratemporal | Variable | |
| PSP | – | PSPS | |
| FTLD-FUS | – | Diffuse localised symmetrical | Early onset bv-FTD |
FTLD-TDP subtypes 1-3 are designated according to Mackenzie's classification.
CBD: corticobasal degeneration; FTD-MND: frontotemporal dementia associated with motor neuron disease; bv-FTD: behavioural variant of frontotemporal dementia; FTLD-FUS: frontotemporal lobar degeneration associated with fused-in sarcoma protein; FTLD-TDP: frontotemporal lobar degeneration associated with TAR DNA-binding protein 43; FTLD-Tau: frontotemporal lobar degeneration with tau inclusions; SD: semantic dementia; FUS: fused-in sarcoma protein; MAPT: microtubule associated protein; PGRN: progranulin; PiD: Pick disease; PSP: progressive supranuclear palsy; CBS: corticobasal syndrome; PSPS: progressive supranuclear palsy syndrome.
Based on data from Rohrer et al.94
It is usually easy to distinguish between FTD and AD based on clinical observation. However, some patients with FTD initially exhibit marked changes in episodic memory, and patients with Alzheimer disease may also show atypical disease presentations. Examples include language-variant AD (usually logopenic PPA), frontal-variant AD (with striking behavioural and executive changes), posterior cortical atrophy (usually presenting with visuospatial and/or visuoperceptual changes and sometimes known as ‘visual variant’), or CBS associated with AD.
Structural MRI with voxel-based morphometry is a useful tool for distinguishing between FTD and AD. FTD exhibits atrophy in the frontal, insular, anterior cingulate, and striate regions, whereas AD shows posterior parietal and occipital atrophy.95 Regional cortical thickness analysis is also helpful, since AD cases will show cortical thinning at the parietal and precuneal levels.96
Other MRI techniques used in distinguishing AD from FTLD include diffusion tensor imaging sequence97 (decreased fractional anisotropy in frontal regions in FTLD), arterial spin labelling98,99 (hypoperfusion in parietal regions and posterior cingulate in AD vs frontal lobe hypoperfusion in FTLD), and combined techniques using positron-emission tomography (PET) with F18-fluorodeoxyglucose (FDG)/RM.100
Several studies performed on atypical variants of AD have shown that, regardless of clinical presentation, AD is associated with impairment in the posterior cingulate gyrus, precuneus, posterior parietal regions, and medial temporal regions.101,102 Other studies have compared patients with PPA and AD to those with PPA only. They found that the patients with AD (usually associated with logopenic PPA) presented more pronounced temporoparietal atrophy, while those with FTLD had imaging studies showing typical ‘knife-edge’ atrophy of the temporal lobe.103,104
Less frequently, FTLD may be confused with Lewy body dementia (LBD), fundamentally in patients who experience visual illusions or hallucinations.105,106 A recent study in a small sample suggested that I123-MIBG scintigraphy may be useful for distinguishing between these 2 entities, because LBD, unlike FTLD, shows a pronounced decrease in contrast uptake.106 In another small study, MR images were not useful for differential diagnosis.107
Functional neuroimaging: cerebral positron-emission tomography and single-photon emission computed tomographyRecent years have seen increasing use of functional neuroimaging techniques, including single-photon emission computed tomography (SPECT) using Tc99m-hexamethylpropyleneamine oxime, or 18F-FDG-PET, in the diagnosis of bv-FTD. Hypoperfusion/hypometabolism is observed in frontal regions in bv-FTD, whereas AD displays hypoperfusion in the temporoparietal and posterior cingulate areas.108,109 Both SPECT and FDG-PET are more sensitive than brain MRI for detecting the early changes occurring in bv-FTD.110 FDG-PET is also very important for detecting phenocopies, since these cases will show preserved metabolism in the frontal regions. However, using FDG-PET in patients whose MRI shows clear cerebral atrophy is unlikely to provide any additional benefit.
Some PET techniques employ new radioisotopes. One uses a compound called carbon 11-labelled Pittsburgh compound B, which binds to β-amyloid and has shown promising results for differential diagnosis of AD and FDT,111,112 especially in cases displaying language alteration.113,114 Other techniques are currently being developed, including PET imaging of tau, PGRN, or FUS. These promising diagnostic tools may be ready for use in the near future.
Biomarkers in plasma and cerebrospinal fluidLow CSF levels of the 42 amino acid beta-amyloid peptide (A β42), and high levels of tau protein, are biomarkers that are both sensitive and specific for diagnosis of AD.115,116 Tau/beta amyloid ratio: A β42 is a useful parameter for distinguishing FTLD from AD since it is lower in FTLD.117 Researchers have reported high levels of TDP-43 in cerebrospinal fluid from patients whose diagnosis of FTD-MND had not been confirmed by a histopathological study.118
Various studies have shown that patients with mutations in the PGRN gene display very low plasma levels of PGRN.54,55,119 Measuring plasma PGRN levels has been proposed as a sensitive and specific screening method for use in these patients.120
Other research indicates that plasma levels of TDP-43 may be related to cerebral impairment in patients with FTLD.121
ConclusionsPicturing FTLD on the molecular level is a way of gaining a more precise understanding of the disease in terms of its causal mechanisms. This also opens the doors to new treatment options. Ligand-specific drugs are currently being developed to search out defined molecular targets.
Of all of the clinical variants, bv-FTD is the most heterogeneous variety, which makes it difficult to predict the underlying disease type. It is possible that this syndrome describes a concept that is too broad and actually includes a number of very different entities. As a result, identifying differential clinical features and defining clinical subtypes may provide better clinical-pathological correlations. In any case, as a complement to clinical evaluation techniques, doctors must develop tools that will deliver in vivo histopathological and molecular diagnoses of these processes.
Structural and functional neuroimaging, biomarkers in laboratory analyses, and genetic studies are being transformed into useful tools for diagnosis or differential diagnosis and for predicting the underlying pathological process. Of all of the biomarkers that have been listed, we should stress the importance of functional neuroimaging. It is more sensitive than structural neuroimaging in early stages of the disease, and it can be used in novel techniques with specific radiotracers (11C-labelled Pittsburgh compound B and tracers for tau, PGRN, and FUS). This technique is emerging as an instrument that promises good results in the near future.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Fernández-Matarrubia M, Matías-Guiu JA, Moreno-Ramos T, Matías-Guiu J. Demencia frontotemporal variante conductual: biomarcadores, una aproximación a la enfermedad. Neurología. 2015;30:50–61.
