



Thrombotic thrombocytopenic purpura (TTP) is a disease with a high rate of mortality if a proper treatment is not instated. Plasmapheresis with plasmatic exchange is the treatment of choice. Diagnosis is performed demonstrating microangiopathic hemolytic anemia, a negative direct Coombs test and thrombocytopenia. Among the clinical data, neurological and renal alterations stand out. When there is a reasonable suspicion in the diagnosis, plasmapheresis must be initiated immediately. There are different diseases that may be similar to the TTP signs and symptoms, especially in pregnant women. TTP has a high risk of relapse and may leave sequelae.
Eli Moschowitz was the first to describe TTP, when performing the autopsy of a 16-year-old girl whom he had treated. During this autopsy, Moschowitz found hyaline thrombi occluding the arterioles and capillaries, he later described them as “aggregated platelets”.1 The presence of hemolytic anemia, negative Coombs, schistocytes, thrombocytopenia, and high lactate dehydrogenase (LDH) in patients with neurological and/or renal alterations9 strongly suggests a TTP diagnosis.
TTP has a mortality rate of 90% without treatment.2 The first attempts to reduce mortality involved large doses of steroids.3 The following attempt included the use of fresh blood.4 Undoubtedly, the turning point in TTP treatment was the use of plasma, which first took place in 1977.5 One of the greatest progresses concerning the comprehension of the physiopathology of this disease was to identify the unusually long von Willebrand factors multimers,6 and later, to identify the deficiency of the protease in charge of splitting this multimers in congenital TTP,7 and lastly, to identify that protease as the thirteenth member of the ADAMTS family (a disintegrin and metalloproteinase with thrombospondin-1 repeats).8
DefinitionThrombotic thrombocytopenic purpura (TTP) is a pathology which puts the patient's life in danger, with a mortality rate of over 90% when a plasmapheresis with plasmatic exchange treatment is not instated. It may affect multiple systems and organs, the nervous system being the one that is usually the most affected. Secondly, it affects the renal function due to the microthrombus occluding the microvasculature. It is clinically characterized by two main laboratory alterations: thrombocytopenia and microangiopathic hemolytic anemia. Direct Coombs test results are negative and LDH is invariably increased.
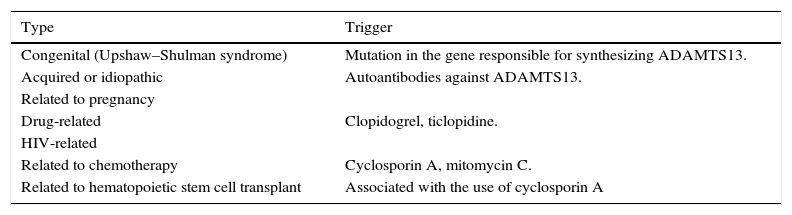
EtiologyTTP is classified according to its etiology (Chart 1), although TTP can be congenital, as a result of a mutation of the gene responsible for desynthesizing ADAMTS13.10 The most common form of TTP is idiopathic or autoimmune, where antibodies directed against ADAMTS13 are created.11
Pregnancy is among the causes of TTP, even though diagnosis in these cases is complicated because the clinical and laboratory findings are very similar to pregnancy-related syndromes like preeclampsia and the HELLP syndrome. In the case of TTP, the signs and symptoms are more severe and the quantification of ADAMS-13 is useful, since in other pathologies it is usually normal.12 Some medications have been linked to the appearance of TTP, quinine being the main responsible.13 Clopidogrel, ticlopidine, cyclosporine A and mitomycin C, among others, have also been linked as TTP triggers.14
Chemotherapy, hematopoietic stem cell transplants, HIV, and lupus are factors frequently related to TTP. They are, however, poorly understood. Hematopoietic stem cell post-transplant TTP is linked to the use of cyclosporine A and is usually a severe clinical picture, and it is difficult to make the patient enter remission.15,16
EpidemiologyYoung women are more prone to this disease. In 2005, there was a study published reporting the incidence of TTP and hemolytic uremic syndrome (HUS), using records which included all patients sequentially for whom plasmapheresis treatment is requested by the Oklahoma Blood Institute due to clinical suspicion of TTP or HUS, based on the presence of microangiopathic hemolytic anemia and thrombocytopenia without an apparent etiology.
The incidence rate for all patients with clinical suspicion of TTP-HUS in the Oklahoma records was 11.29 per million inhabitants. The incidence rate for idiopathic TTP-HUS patients was 4.6 per million inhabitants, and the incidence rate for patients with severe ADAMTS13 deficiency was 1.74 per million inhabitants.
The incidence rate of TTP associated with severe ADAMTS13 deficiency is over 9 times higher in black people than other races. The incidence rate of TTP-HUS comparing women with men was >1.17
PhysiopathologyThe main histological finding in TTP is hyaline thrombi, formed by platelet aggregation. These thrombi are responsible for capillary occlusion, with the subsequent ischemia in different organs.1,18
The von Willebrand Factor (vWF) plays a key role in platelet aggregation. VWF produced exclusively in endothelial cells and platelets, is stored as Wiebel-Palade granule in endothelial cells and as α-granule in platelets. Within these granules it is stored in an ultra-large shape (ULvWF).7 After being secreted, ULvWF attaches to the endothelial surface. The longer the vWF is, the greater the effect on platelet aggregation, because it is more related to the platelet's Ib receptor. The role of ADAMTS13 is to split the ULvWF into smallest molecules.9,19 In congenital TTP, ADAMTS13 activity may be lower than 5%, due to mutations in the gene responsible for ADAMTS13 production.10 Regarding idiopathic TTP, findings show that it is an autoimmune process, mediated by IgG-class antibodies.11 Even though ADAMTS13 deficiency is suggested as the main culprit, it is important to mention the fact that other causes have been studied, such as lymphocytes, macrophageactivation, high IL-1, IL-6, IL-2 and TNF-α levels, and transforming growth factor (TGF)-β, among others.20,21
Signs, symptoms and laboratory findingsClassically, a pentad is described, consisting of thrombocytopenia, microangiopathic hemolytic anemia, fever, neurological symptoms and renal dysfunction.2 It is worth stressing that currently this pentad is rarely seen, since treatment with plasmapheresis is instated before it occurs. Nowadays, the only thing required is thrombocytopenia and microangiopathic hemolytic anemia in order to establish diagnosis, and, as mentioned above, the presence of schistocytes, a negative direct Coombs and a high LDH confirm clinical suspicion of TTP (Tables 1 and 2).
Classification of TTP.
| Type | Trigger |
|---|---|
| Congenital (Upshaw–Shulman syndrome) | Mutation in the gene responsible for synthesizing ADAMTS13. |
| Acquired or idiopathic | Autoantibodies against ADAMTS13. |
| Related to pregnancy | |
| Drug-related | Clopidogrel, ticlopidine. |
| HIV-related | |
| Related to chemotherapy | Cyclosporin A, mitomycin C. |
| Related to hematopoietic stem cell transplant | Associated with the use of cyclosporin A |
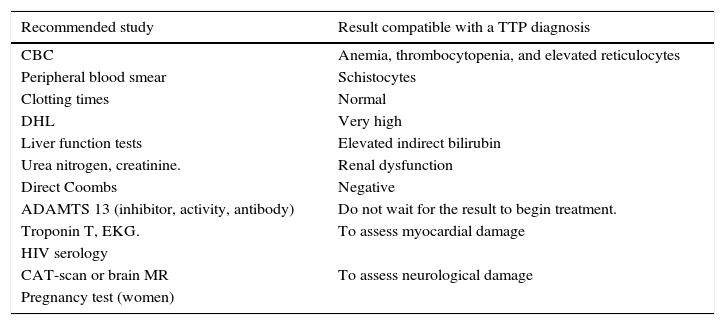
Recommended studies for patients with a suspected TTP diagnosis.30
| Recommended study | Result compatible with a TTP diagnosis |
|---|---|
| CBC | Anemia, thrombocytopenia, and elevated reticulocytes |
| Peripheral blood smear | Schistocytes |
| Clotting times | Normal |
| DHL | Very high |
| Liver function tests | Elevated indirect bilirubin |
| Urea nitrogen, creatinine. | Renal dysfunction |
| Direct Coombs | Negative |
| ADAMTS 13 (inhibitor, activity, antibody) | Do not wait for the result to begin treatment. |
| Troponin T, EKG. | To assess myocardial damage |
| HIV serology | |
| CAT-scan or brain MR | To assess neurological damage |
| Pregnancy test (women) |
Clinical findings which pave the way to thrombocytopenia suspicion are epistaxis, menorrhagia, hematuria, gingivorrhagia, etc. Nevertheless, significant hemorrhage data is not necessarily found, as in the case of autoimmune thrombocytopenic purpura. Platelets are below normal levels, typically less than 20×109/L. Lab work shows hemoglobin below normal levels, elevated reticulocytes, elevated indirect bilirubin, elevated LDH, low haptoglobin, normal coagulation time, usually in early stages of the disease, and a negative Coombs direct test can also be used. In a peripheral blood smear we are able to observe schistocytes or crenocytes due to their mechanical fragmentation when going through occluded capillaries.22,23
Regarding nervous system symptoms, in summary these are the focal symptoms: cephalalgia, paresis, aphasia, cognitive deterioration, and transitory ischemic accidents. Renal dysfunction is not as marked as hemolytic uremic syndrome, and rarely requires dialysis,24 though proteinuria and oliguria, among others, may appear. It is important to take cardiac affection into consideration. This is typically under diagnosed, and may occur in acute infarction, congestive cardiac failure, arrhythmias, cardiogenic shock, and even sudden death.25,26 The disease's natural course without treatment leads to death for 90% of patients in a matter of days.27
Differential diagnosesHemolytic uremic syndrome (HUS)It is usually preceded by a period of hemorrhagic enterocolitis caused by the shiga toxin, produced by diverse serotypes of E. coli (i.e. 0157:H7) or Shigella spp., particularly in children. HUS is a disorder in the activation of a complement, in order to diagnose it, it is necessary to prove the presence of the shiga toxin and for the complement to be depleted. There are two types of HUS, the typical type which occurs in children and the patient usually recovers on his/her own, and the atypical, which is most common in adults. Sometimes there is no diarrhea or a clear trigger and the patient's course with renal failure, which in most cases evolves into renal failure that require dyalisis.24,28
PreeclampsiaIt develops in the 2nd or 3rd trimester, with the onset of hypertension and proteinuria after week 20 of pregnancy. Less than 5% of women with preeclampsia will develop severe thrombocytopenia.12
HELLP syndromeHemolysis, Elevated Liver enzymes and Low Platelets (HELLP). A key point in the diagnosis of this pathology is that ADAMTS13 activity is normal. Lab work shows an elevation of LDH, AST, and thrombocytopenia. A LDH/AST ratio >22.12 suggests that a TTP diagnosis is more probable than HELLP syndrome in pregnant patients during their third trimester.12,29
Diagnostic methodsDiagnosis is performed through clinical history and the examination of a peripheral blood smear. The measurement of ADAMTS13 levels and anti ADAMTS13 antibodies is recommended; however, plasmapheresis and plasma exchange treatment should not be delayed while waiting for the results of these measurements. The requirements to begin plasmapheresis are merely the presence of schistocytes in peripheral blood (more than 2 per field), anemia, thrombocytopenia and elevation of LDH.
As previously pointed out, the classic pentad of clinical signs and symptoms is no longer considered current; nowadays the presence of thrombocytopenia and microangiopathic hemolytic anemia is enough to create the suspicion of a TTP diagnosis. The recommended laboratory tests to request (chart 2) are useful in order to confirm diagnostic suspicion of TTP, rule out differential diagnoses and to assess white organ damage.30
Current treatmentPlasmapheresis with plasma exchange is the treatment of choice. This therapy should begin immediately if there is the suspicion of TTP.
Plasmapheresis has two main objectives:
- •
Withdrawal of the ADAMTS13 inhibitor (IgG), as well as most of the ULvWF chains.
- •
Replacement of deficient or missing ADAMTS13.
Plasmapheresis is superior to plasma infusion.31 However, it is possible to use a high dose of plasma infusion (20–30ml/kg/day) as an initial therapy, especially when there is no access to resources to begin plasmapheresis, and even though there is the risk of a volume overload and proteinuria exacerbation. Plasma infusion is not a substitute for plasmapheresis; it just functions as a bridge therapy.32
The exact duration of treatment and the necessary number of sessions is highly variable between patients, depending on the presence or the title of the autoantibody and ADAMTS13 activity.
British guidelines establish plasmapheresis to be started with 1.5 times plasma volume for 3 days, and reassess the patient daily. They recommend the use of plasma treated with solvent/detergent where with the risk of infections and adverse immune responses decreases.30,33 The exchange volume may be lowered to 1 if there is clinical and lab improvement. Similarly, it may be increased in cases where the patient's life is at risk. At our center plasmapheresis is continued until we obtain a number higher than 150×109/L platelets and normal LDH. After reaching this number, the case is monitored daily before deciding on a higher plasmapheresis sessions. However, in hospitals or places with higher economic resources, it is reasonable to continue with at least 2 plasmatic exchanges once TTP remission has been obtained.
The use of steroids is very controversial. No benefits have been clearly demonstrated, yet they are widely used.
RituximabWhen dealing with autoimmune TTP, it is logical to use an antibody whose targets are the lymphocytes B responsible for the immune response mediated by antibodies. Such is the case with rituximab, whose only objective is the antigen CD20 on the surface of the lymphocytes B.
The use of rituximab has been suggested if there is cardiac or neurological compromise.34 However, its use in all patients from the start is promising. In an acute TTP episode, initial treatment with rituximab, plasmapheresis and corticosteroids have an apparent remission in >90% of the patients within 14 to 21 days and may also reduce relapse frequency.34
In patients with an episode of refractory TTP, the addition of rituximab to plasmapheresis and corticosteroids increases platelet counts in >80% of the patients and may decrease the required time to accomplish a response platelet count.35
A persistent severe deficiency of ADAMTS13 during remission should lead to the consideration of administering rituximab as a preventive measure to avoid relapse.36
In our center, we published 4 cases of TTP treated with low doses of rituximab, plasmapheresis and a short steroid course. Patients were administered 100mg of rituximab intravenously, from the second to eighth day, with the beginning of plasmapheresis as a first line therapy in three cases and as a rescue therapy for relapses in one case. The number of necessary plasmaphereses varied from 5 to 12 sessions. All patients reached complete remission and are currently asymptomatic, with a duration of complete response of 8–22 months.37 We treated 7 more patients for a total of 11 and in data – as yet unpublished – we accomplished a disease-free survival rate of 2 years of 90%. In Fig. 1 we present a scheme showing the therapeutic approach of TTP in different clinical scenarios.
New treatmentsRecombinant ADAMTS13
A study was conducted in order to find out whether or not an ADAMTS13 recombinant is capable of bearing the neutralizing antibodies and restoring ADAMTS13 activity in patients with acquired TTP. The relationship between the amount of added ADAMTS13 recombinant, the recovery of ADAMTS13 activity and the titles of the anti ADAMTS13 inhibitors in the patient was investigated.
They demonstrated that an ADAMTS13 recombinant, at a reasonable dose, has the ability to bear the neutralizing inhibitors and rebuilding ADAMTS13 in vitro activity in acquired TTP.38
PrognosisBefore plasmapheresis, TTP had a mortality rate of over 90%, but with the use of plasmapheresis mortality rate went down to 20%.39 Despite the major advances in the reduction of the mortality rate, patients who survive TTP face a series of challenges: a major morbimortality rate compared to the general population,40 as well as a lower quality of life, less social life and cognitive deficits, in addition to the risk of relapse.41–44 Over a third of patients who survive an acute episode of TTP will have at least a relapse during the following 10 years.43 Relapse rate is 34%, most initial relapses among patients with ADAMTS13 activity <10% occur within the first year after remission and this rate decreases with time.44
Conflicts of interestThe authors have no conflicts of interest to declare.