



La hipertensión portopulmonar (POPH), es una entidad clínica relativamente frecuente en sujetos con enfermedad hepática avanzada. Esta enfermedad se caracteriza por un aumento en la presión de la arteria pulmonar y en la resistencia vascular pulmonar que finalmente desencadena falla ventricular derecha. La presencia de esta condición en candidatos a trasplante de hígado acarrea un impacto negativo en la sobrevida, por lo que deben hacerse esfuerzos para la detección y tratamiento oportuno. Este tópico será revisado aquí enfatizando en el tamizaje, tratamiento y el papel del trasplante de hígado en esta enfermedad.
Portopulmonary hypertension (POPH,) is a relatively frequent entity in subjects with advanced liver disease. This disease is characterized by an increase of the pulmonary artery pressure and the pulmonary vascular resistance that finally does cause right ventricle failure. The presence of this condition in liver transplant candidates carries a negative impact on survival, efforts should be done for early detection and treatment in a timely manner. This topic will be reviewed here, emphasizing on the screening, treatment and the role of liver transplantation in this disease.
Introduction
Pulmonary vascular complications associated with portal hypertension (cirrhosis) are hepatopulmonary syndrome (HPS) which is characterized by intrapulmonary vascular dilatations, and portopulmonary hypertension (POPH) characterized by an increase in pulmonary artery pressure and resistance. This paper will focus on the latter.
The first description of POPH was published in 1951; Mantz and Craige described the case of a 53 years old female with portal hypertension secondary to a large portocaval shunt and an enlarged pulmonary artery on autopsy. Microscopic examination of the lungs showed multiple clots on distal arterial vessels as well as intimal thickening.1 POPH is diagnosed when a patient with portal hypertension has evidence of pulmonary arterial hyper-tension (PAH) confirmed by a right heart catheterization (RHC) and other causes of pulmonary hypertension have been excluded such as congenital heart disease, thromboembolic disease, connective tissue disease, untreated sleep apnea, interstitial lung disease, HIV and drugs.2
The criteria for POPH diagnosis are the presence of portal hypertension and:
- Mean pulmonary artery pressure (mPAP) >25mmHg.
- Mean pulmonary artery occlusion pressure (mPAOP) <15mmhg and if the mpaop is 15mmHg, a transpulmonary gradient (TG = mPAP - mPAOP) >12mmHg.
- Pulmonary vascular resistance (PVR) >240 dynes.s.cm-5.
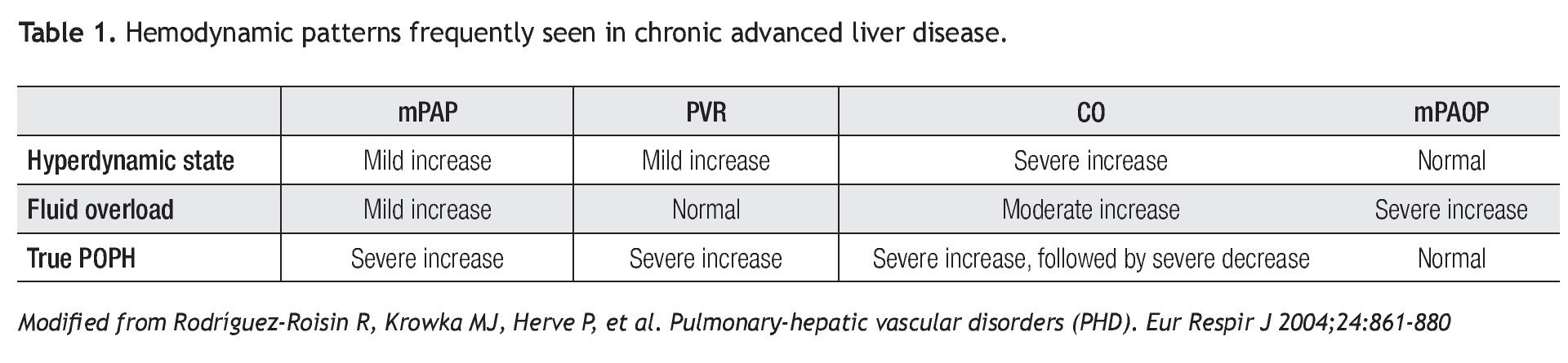
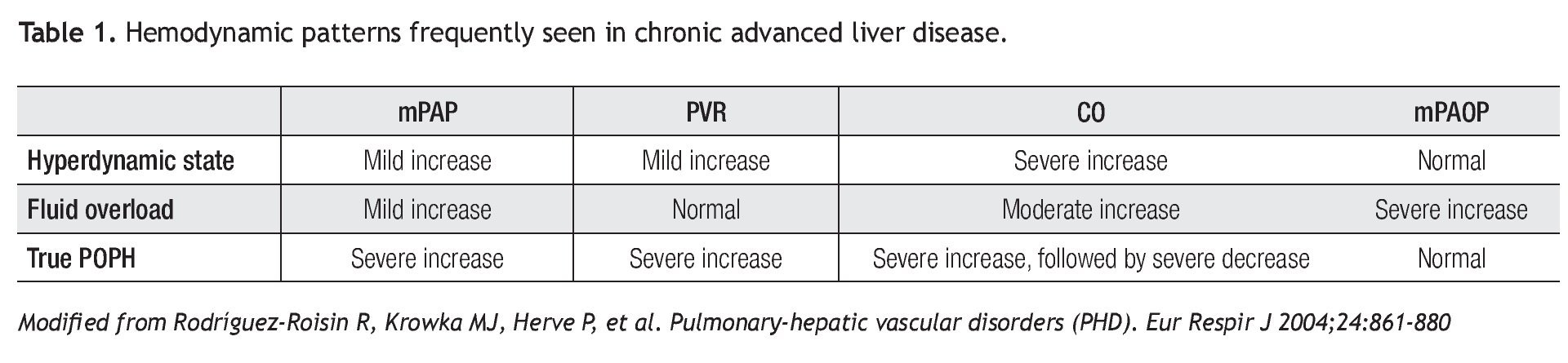
Between 30% to 50% of patients with liver disease have an increased cardiac output (CO) and a mild to moderate elevation in mPAP without the features of portopulmonary hypertension.3,4 In addition, these patients often have volume overload and have high mean PAOP. Other causes related to an increase of pulmonary artery pressures are cirrhotic cardiomyopathy and intrahepatic arteriovenous fistulas.5 As conditions of fluid overload and high CO can also contribute to pulmonary hypertension, it is important to recognize the variable RHC hemodynamic patterns in patients with liver disease (Table 1).
The World Health Organization classifies pulmonary hypertension into groups with similar underlying pathology and shared therapeutic managements. On group I, POPH is included along with idiopathic pulmonary artery hypertension (IPAH), heritable pulmonary hypertension and pulmonary arterial hypertension associated with connective tissue disorders, congenital systemic-to-pulmonary shunts, HIV infection and drugs.6
EpidemiologyAutopsy series in cirrhotic patients have found pathologic changes consistent with PAH in 0.7% of the cases.7 It is difficult to estimate the true prevalence or incidence of POPH since diagnostic criteria were not homogeneous in earlier studies and most of the data regarding to this issue was obtained from studies of patients with advanced liver disease, or in liver transplant (LT) candidates. The prevalence of POPH in patients with chronic liver disease undergoing to a LT is between 4% and 8%,8,9 while on patients with refractory ascites, it is observed in 16% of cases.10
Pathogenesis
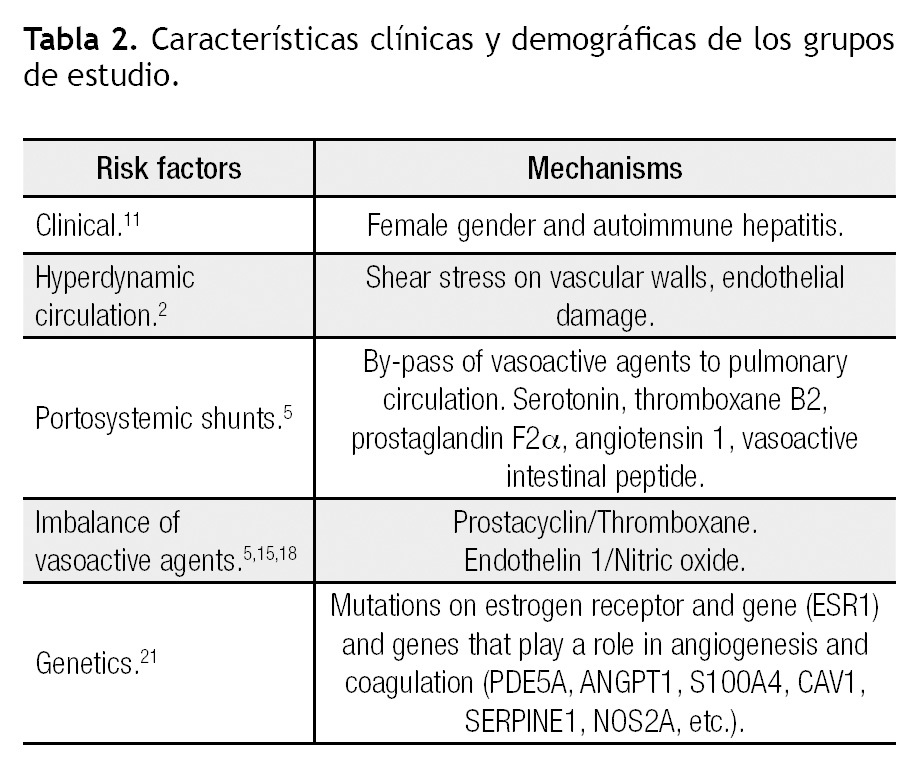
Portal hypertension is required for the development of POPH. Conditions that can cause portal hypertension and thus are implicated in the etiology of POPH include cirrhosis (most common), portal vein thrombosis, hepatic vein sclerosis, congenital portal circulation abnormalities, biliary atresia and periportal fibrosis without cirrhosis. Systemic conditions that may involve portal and pulmonary vessels are antiphospholipid syndrome, mixed connective tissue disease, schistosomiasis, sarcoidosis, systemic lupus erythematosus, microangiopathic hemolytic anemia and HIV.5 why patients with portal hypertension develop POPH is unknown. Several mechanisms have been associated, but at this moment the exact trigger for developing pulmonary hypertension is yet to be discovered (Table 2). The Pulmonary vascular Complications of Liver Disease Study Group (PVCLD) reviewed the clinical data in patients with POPH and found two clinical risk factors for the development of PAH; female gender (adjusted odds ratio = 2.90, 95% confidence interval 1.20 - 7.01, p = 0.018) and autoimmune hepatitis (adjusted odds ratio = 4.02, 95% confidence interval 1.14 - 14.23, p = 0.031). This suggests that hormonal and immunologic mediators may play a key role in the genesis of POPH. Of note, patients with Hepatitis C related cirrhosis have a lower risk of developing POPH (adjusted odds ratio = 0.24, 95% confidence interval 0.09 - 0.65, p = 0.005).11
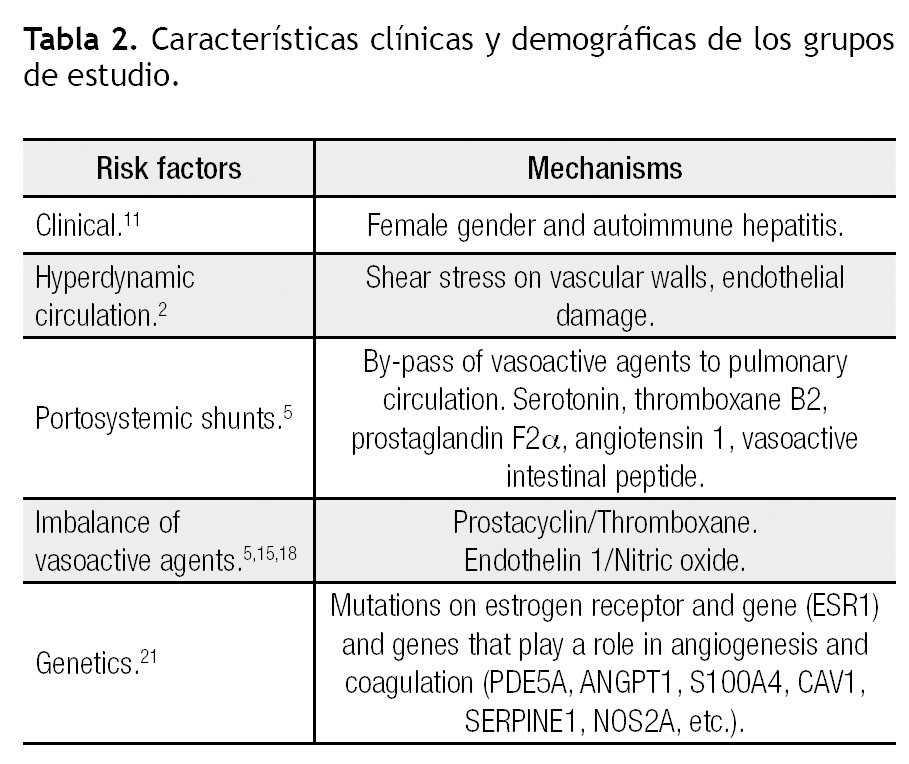
The hyperdynamic circulation seen in patients with liver disease has been postulated to contribute to the development of POPH.12 The increased blood flow through the pulmonary vascular bed and increased shear stress on the vascular wall may damage the endothelial cells and lead to the arteriopathy seen in PAH. However, this mechanism remains speculative and currently lacks supportive data.
A popular hypothesis in the pathogenesis of POPH is that of humoral mediators escaping from the hepatic metabolism and reaching the pulmonary circulation through portosystemic collaterals. Substances like endothelin-1, serotonin, thromboxane B2, prostaglandin F2α, and vasoactive intestinal peptide are increased in patients with POPH. Serotonin has been identified as a potential mediator in the pathogenesis of IPAH through its vasoconstrictive and proliferative effects in pulmonary vessels in vivo.13 In normal conditions, the liver metabolizes serotonin before it reaches the pulmonary circulation. when liver dysfunction exists or there are portosystemic collaterals, the pulmonary circulation can be exposed to high levels of serotonin.3,5
Imbalance of endothelium-derived vasoactive mediators has been the focus of an alternate hypothesis for the pathogenesis of POPH. Prostacyclin is produced in the vascular endothelium and smooth muscle cells. It is a potent vasodilator, inhibits platelet adhesion and opposes hypoxia-mediated vasoconstriction. Prostacyclin also has antiproliferative properties on the pulmonary arterial smooth muscle cells.14 Thromboxane B2, a stable meta-bolite of thromboxane A, has potent vasoconstrictor and procoagulant properties. A study in patients with IPAH and secondary causes of PAH found an increased urinary excretion of thromboxane B2 and a decreased excretion of prostacyclin in patients with PAH compared with normal and COPD subjects.15 Also a decreased expression of
prostacyclin synthase in small and medium sized pulmonary arteries of patients with pulmonary arterial hypertension has been demonstrated.16 Endothelin-1 (ET1) is a potent vasoconstrictor with mitogenic activity in pulmonary arterial smooth muscle cells implicated in the pathogenesis of PAH.17 Patients with cirrhosis have plasma ET1 concentrations three times higher than healthy controls. ET1 may also be involved in the direct pathogenesis of portal hypertension.10 Endothelial dysfunction associated with reduced nitric oxide (NO) is another potential pathogenic mechanism in PAH.18
Potential role of genetic mutations in the development of POPH are still under investigation. Although mutations of the bone morphogenetic protein receptor-2 (BMPR-2) has been linked to heritable pulmonary arterial hypertension and IPAHs,19,20 this mutation has not been associated with POPH.3,21 The PVCLD genotyped 1,079 single nucleotide polymorphisms (SNPs) in 93 candidate genes in an attempt to identify common genetic variations associated with the risk of POPH in a group of patients undergoing LT evaluation.21 They found 29 SNPs in 15 genes related to POPH. It should be noted that there were 7 SNPs in the estrogen receptor 1. Other genes with SNPs related to POPH were the aromatase, phosphodiesterase 5, angiopoetin 1, calcium binding protein A4, caveolin 1, xantine deshydrogenase, superoxide dismutase 2, NADPH oxidasa 4, plasminogen activator inhibitor 1, nitric oxide synthase 2A, retinoic acid receptor beta, homolog of Drosophila mothers against dpp3, runt-related transcription factor 1 and recombining binding protein 1 for J-kappa. These genes are involved in angiogenesis, control of coagulation and vascular tone pathways.
The histopathological findings of the pulmonary vessels of POPH are indistinguishable from other forms of PAH. Intimal proliferation and/or thickening, medial smooth muscle hypertrophy and fibrosis are seen in the small pulmonary arteries. In situ thrombosis with recanalization may also be present. The plexiform lesion, which is a dilated pulmonary artery with the normal structure replaced by an intraluminal plexus of endothelial cells and slit-like vascular channels, is also found in POPH.22
Clinical findings
In POPH, patients have clinical evidence of both portal hypertension and PAH. But in the early stages of POPH, patients may have no symptoms or only symptoms of liver disease. Rarely, they do present with respiratory complaints related to PAH without clinical evidence of portal hypertension. Patients with portal hypertension who report dyspnea at rest or during exercise must been screened for POPH.23 Chest discomfort and syncope are features of advanced POPH. Physical examination may show an elevated jugular venous pressure, an accentuated P2 component, a tricuspid regurgitation murmur, right ventricular heave or increasing lower extremity edema. Other signs of right ventricular failure include the presence of right sided S3 and the presence of a pulsatile liver.
Chest radiographs typically demonstrate prominent pulmonary arteries and cardiomegaly without relevant parenchymal abnormalities. The electrocardiogram (ECG) can demonstrate right atrial enlargement, right ventricular hypertrophy, right axis deviation and a right bundle branch block. Pulmonary function test may show a decreased diffusion capacity of carbon monoxide (DLCO), but serves primarily to screen for underlying respiratory disorders.3
Screening
All patients undergoing evaluation for liver transplantation should be screened for POPH. This condition increases the risk of perioperative death and should not be diagnosed in the operating room24. Patients with portal hypertension and dyspnea also should be screened for possible POPH.
Transthoracic Doppler echocardiography (TTE) is the screening test of choice for POPH23. The echocardiographic parameter used for screening is the right ventricular systolic pressure (RVSP), which is calculated from the peak tricuspid regurgitant velocity (TRV) and an estimate of the right atrial pressure (RAP), using the modified Bernoulli equation (RVSP = 4 (TRV)2 + RAP). Using a cutoff of RVSP ≥50 mmHg, TTE has a sensitivity of 97% and a specificity of 77%.25 Although, TTE confidently identifies patients with moderate to severe POPH (mPAP >35 mmHg), it is not specific enough and does not characterize severity. Other caveats of TTE are interobserver variability, use of estimations for the calculation of RAP, and inability to detect a measurable tricuspid regurgitant flow (TRF) in 18 to 68% of screened patients.4,25,26 when it is not possible to detect a measurable TRF, clinical information and other signs of PAH in TTE such as enlargement of right heart chambers and septum displacement should be consider in the decision to proceed to RHC. Considering the factors mentioned above, all liver transplant candidates with a RVSP ≥50 mmHg, or with symptoms of dyspnea or other clinical signs of pulmonary hypertension, should undergo to a RHC to characterize the pulmonary hemodynamics.
Right heart catheterizationAs previously mentioned, the diagnosis of POPH relies on RHC derived parameters. RHC can accurately measure mPAP, PAOP (to exclude volume overload), cardiac output (to exclude high-output pulmonary hypertension), and PVR (Table 1). In addition, hepatic vein wedge pressure (HvwP) and free hepatic vein pressure (FHVP) can be measured to confirm portal hypertension. A hepatic vein pressure gradient (HvwP-FHVP) greater than 4 mmHg indicates the presence of portal hypertension.27 These pressures can be measured readily from the femoral vein approach without the need for additional guide wires.
Vasodilator studies with NO, epoprostenol or adenosine are not routinely done in this group of patients. In IPAH, a positive response to vasodilators (decrease of mPAP of 10 mmHg to a value below 40 mmHg) confers a better prognosis. Those patients can be treated with calcium channel blockers (CCB) with good tolerance and response.28 In POPH, therapy with CCB usually is not tolerated or contraindicated because they can exacerbate edema and portal hypertension. A positive vasodilator response in POPH does not predict survival.3
Approximately 30% to 40% of patients with RVSP ≥50 mmHg by TTE have a hyperdynamic and/or fluid overload pattern on RHC.4,29 This group of patients does not meet the criteria for POPH and do not warrant aggressive treatment with PAH specific therapies. As their risk for LT is not increased,24 LT should not be delayed for these patients.
TPG calculation is relevant in patients with elevated PAOP (fluid overload). When a gradient of more than 12mmHg is found in association with increased PVR and mPAP, the pulmonary hypertension may have pre and/or post-capillary components.
When POPH is confirmed with RHC, the next step is to classify in mild, moderate or severe POPH according to the mPAP value (Figure 1). In addition characterizing the hemodynamic pattern, RHC results have a significant effect on treatment decisions.
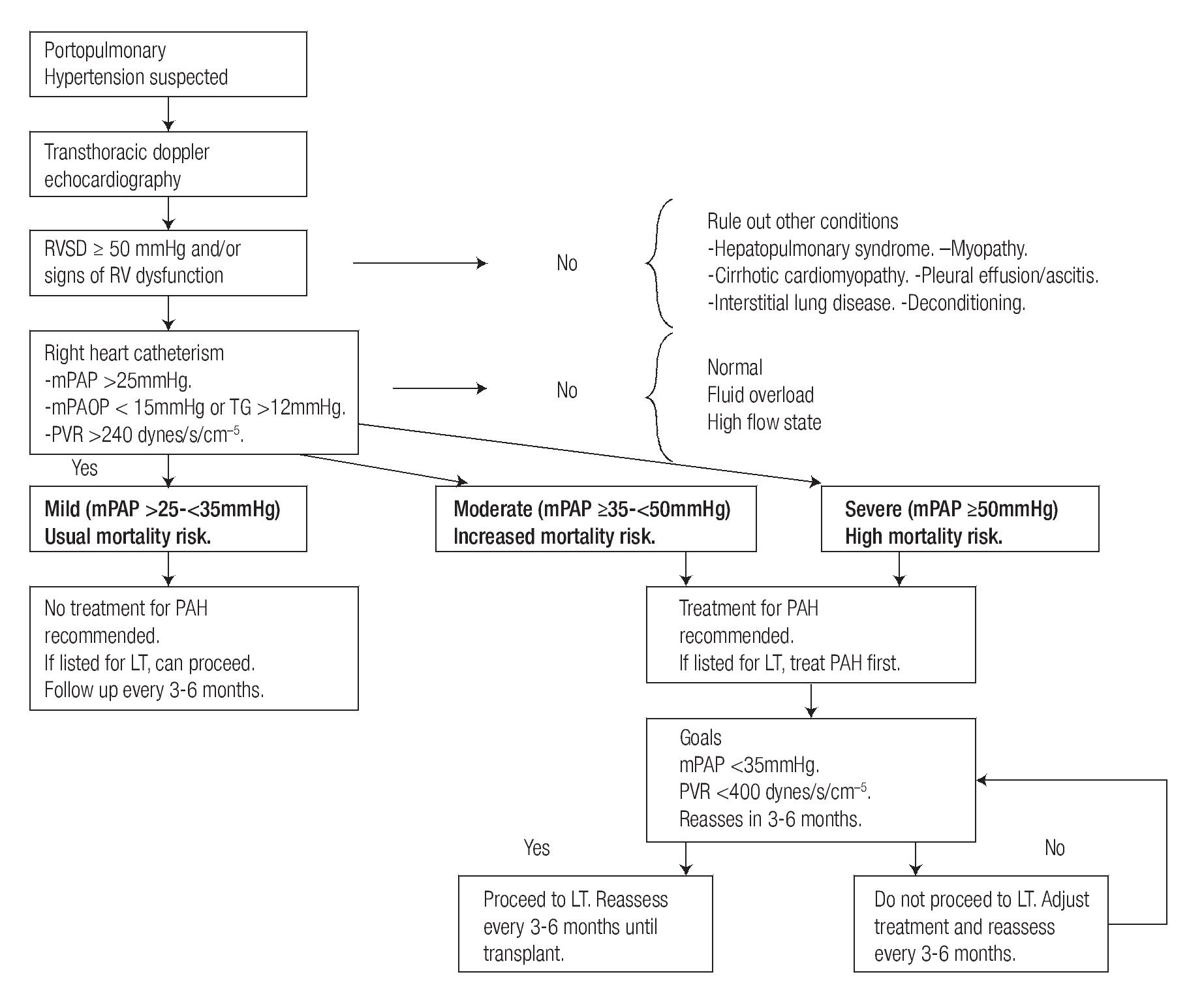
Figure 1. Suggested approach for screening and treatment of patients with POPH.
Treatment
Randomized controlled trials in this group of patients are lacking. Most of the treatment options are extrapolated from trials in patients with IPAH or from case series and small observational studies (Figure 1).
General measures. Anticoagulation is usually contra-indicated in POPH due to the risk of gastroesophageal variceal bleeding and thrombocytopenia. Recent guidelines do not support routine use of anticoagulation therapy in patients with associated PAH.30
Diuretics, as in IPAH, are a mainstay in the treatment of POPH, by alleviating the right ventricle of fluid overload. However, the role of diuretics and POPH it has not been established. Due to concerns of exacerbating hepatic encephalopathy, the use of diuretics needs to be tailored to each patient with awareness of its potential benefits and risks.
Transjugular intrahepatic portosystemic shunts (TIPS) have no role in the treatment of POPH and may acutely increase preload, CO and mPAP. Transient increases of PVR have also been documented. These changes may have detrimental effects on an already dysfunctional right ventricle in the setting of moderate to severe POPH.3
Calcium channel blockers (CCB) usually are the first line of treatment in patients with IPAH who have a positive response to acute vasodilator challenge during RHC, but are contraindicated in POPH. CCB may increase mesenteric flow leading to an increase in portal venous pressure and a widened hepatic venous pressure gradient.2
Patients with portal hypertension often receive non-selective beta blockers for the primary or secondary prophylaxis of variceal bleeding. However, beta blockers are contraindicated in other forms of PAH. whether the routine use of beta-blockers in POPH leads to worsening morbidity or mortality has not been studied. One report found withdrawing beta blockers in patients with moderate to severe POPH led to an increase in CO, decrease in PVR and a better performance in 6 minute walk test (6MwT).31
Pulmonary arterial hypertension specific therapy. Specific therapy for PAH refers to the administration of agents with complex mechanisms of action including vasodilation and effects on vascular growth and remodeling. Three groups of drugs, prostanoids, endothelin-1 receptor antagonists (ERA) and phosphodiesterase 5 inhibitors (PDE5 inhibitors) are currently available for the treatment of PAH.
1. Prostanoids. Currently in the US, there are three agents commercially available: intravenous (IV) epoprostenol, treprostinil (IV, subcutaneous and inhaled) and inhaled iloprost (iILO).
Epoprostenol is a potent vasodilator with antiplatelet and antiproliferative properties shown to improve survival, hemodynamics, exercise tolerance and functional class in IPAH.32 It is administered as a continuous IV infusion via an indwelling central venous catheter, and a portable infusion pump. Experience using IV epoprostenol in POPH is wider than with other agents, especially as a bridge to liver transplant. Early reports showed short and long term effectiveness of epoprostenol reducing PVR, mPAP and increasing CO.33-35 Of note, in one study, 6 of 10 patients on long term epoprostenol died. Three of the deaths were associated with progression of portal hypertension but stable PAH.34 This raised initial safety concerns about using epoprostenol in this condition. while using epoprostenol in POPH, cases of progressive splenomegaly and hypersplenism have been reported. This may be due to increased blood flow to the splachnic circulation.35,36
But more recent reports have shown that epoprostenol is safe and can be used successfully to treat patients with moderate and severe POPH as a bridge to liver transplantation.37-41
Iloprost has the advantage of being given by inhalation, but requires six to nine inhalations per day. Recently, the acute effects on pulmonary and hepatic hemodynamics of iILO were described in 21 patients.42 Using 2.8 μg of iILO, baseline and 1 hour post dose measurements were done. A significative decrease in mPAP (16% ± 8%) and PVR (18% ± 14%) was observed. The HvPG and hepatic blood flow did not vary during the study. The same group retrospectively evaluated 12 patients treated with iILO 30 μg/d for more than one year and they found a significant improvement in functional class and 6MWT. One patient died and another changed treatment in the observation period.42
In a case report of 3 patients using IV treprostinil, two of them had an uneventful liver transplantation. The remaining patient had a mPAP decrease from 60 mmHg to 44 mmHg after two years of treatment (106 ng/kg/min), a significant decrease but not enough to list for a liver transplant.43
Common side effects of prostanoids, especially with intravenous formulas, are jaw pain, diarrhea, flushing and lower extremity pain. For POPH patients with potential for concomitant encephalopathy and/or recurrent bleeding complications, the risk and benefits of embarking on IV continuous prostanoids therapy must be individualized and emphasized. Also, the treatment goals and expectations must be coordinated with the LT team for those patients receiving bridging therapy toward eventual or possible LT.
2. ERAs. Currently there are two agents available in this group, bosentan (dual antagonist of A and B receptor) and ambrisentan (highly selective antagonist of A receptors). In a retrospective study,44 the authors assessed the safety and efficacy of bosentan in 18 patients with Child A and B cirrhosis and severe POPH. The patients had significant improvement in exercise capacity and hemodynamic parameters. More remarkable was the survival rates at 1, 2 and 3 years of treatment of 94%, 89% and 89% respectively compared to the mortality rate for this disease if left untreated that is between 24% and 60% in the first year. Also in this study, there was a subgroup of 13 patients treated with iILO only. They also had good response in exercise capacity and hemodynamic parameters but survival rates at 1, 2 and 3 years were inferior compared to those treated with bosentan (77%, 62% and 46% respectively). In another study that included 11 patients with POPH treated with bosentan for a year, significant sustained improvements were seen after a year of treatment in PVR, cardiac index (CI), vO2max (maximal oxygen consumption), and 6MwT.45 In the pivotal BREATHE-1 study,46 11% of patients with PAH had elevations in liver function tests (LFTs). Although in the previously cited studies there were no significant increases of liver enzymes or hepatic failure related to bosentan, there are four cases reported of severe liver toxicity related to bosentan in POPH patients.47 Ambrisentan is a highly selective ERA antagonist (type A receptors), with fewer cases of reported liver function tests elevation in the two pivotal studies.48 Recently a cohort of patients with POPH treated with ambrisentan,49 showed significant improvement in mPAP, PVR, CI, and wHO functional class from the baseline to the period of evaluation (≤15 months). No patients in this study had alterations in the LFTs. In spite of these reports of benefit in POPH, both agents are not recommended for patients with moderate to severe liver disease. Accordingly, the routine use of ERA in POPH cannot be endorsed.
3. PDE5 inhibitors. In a report of 14 patients (1 moderate, 13 severe POPH) treated with sildenafil,50 8 were PAH treatment naive and 6 were previously on inhaled prostanoid (iloprost n = 5, treprostinil n = 1). They reported significant increase in 6 MWT and decrease in PVR and pro-BNP. These improvements where sustained for 12 months. A significant reduction in mPAP was observed at three months compared to baseline, but not at 12 months. Studies in animal models using sildenafil and vardenafil show an increased flow in portal vein flow in cirrhotic rats without an increase in portal pressure.51 In a study, 18 healthy subjects and 18 with Child A cirrhosis received a single oral dose of 10 mg of vardenafil5. Doppler sonographic measurements of the hepatic artery and splachnic flow were perfomed at 1 hour and 48 hours after vardenafil dose. An increase of portal vein flow was observed; in 4 of the 5 patients, the transportal gradient by catheterization also decreased.52 Tadalafil has also been shown to decrease mPAP and portal pressure.53 Accordingly, the combination of favorable changes to the portal pressure as well has pulmonary hemodynamics makes PDE5 inhibition an appealing target in POPH. However, as with the other current PAH specific agents, RCT data in POPH is lacking.
Liver transplantation
Traditional thinking had been that POPH is a contraindication for LT. Currently we look to LT as being a potentially curative intervention for select patients with POPH. In patients who successfully undergo LT, normalization or improvement of pulmonary hypertension can take months or even years. This may be related to the degree of pulmonary vascular remodeling prior to LT. There are reports of progression, recurrence and even de novo PAH after LT.54,55
Careful selection of POPH patients for LT is paramount. The risk stratification for LT focuses on the degree of PAH and right ventricle function. One LT center published their experience that showed intraoperative mortality rates in mild (n = 14), moderate with PVR <250 dynes s cm- 5 (n = 6), moderate with PVR >250 dynes/s/ cm- 5 (n = 14), and severe POPH (n = 6) of 0%, 50% and 100% respectively. 24 Moderate POPH with raised PVR and severe POPH are associated with a worse prognosis if they undergo LT without proper evaluation and treatment. In the latter study TPG ≥15 mmHg was seen in 42% of the patients who died and no deaths were observed in patients with TPG <15 mmhg interestingly right ventricle failure was the cause of death in most cases untreated patients median survival poph is approximately 18 months 56 But as mentioned above, moderate and severe POPH give unacceptable high perioperative mortality to the LT procedure. There is a proposal to give a high priority for LT to POPH patients who responded favorably to PAH therapy. 57
According to experts recommendation after PAH specific therapy has been initiated, follow up RHC is recommended for every 3 to 6 months, pursuing as a goal a mPAP ≤35 mmHg and PVR <400 dynes s cm- 5 Taking into account that patients die from Rv failure, some authors recommend testing Rv function with an acute volume challenge after the hemodynamic goals are reached with medical therapy. 58 Epoprostenol is the most studied drug to treat patients with severe POPH who are candidates for LT. A recent report 39 describes a group (n = 8) of patients who had POPH as their only contraindication for LT, and they were treated with epoprostenol. Seven patients responded favorably decreasing mPAP below 35mmHg, six of them within 6.5 months and one 14 months after treatment. They were classified as either early or late responders. The early responders were activated in the transplant list, four of them underwent to LT successfully. Epoprostenol was discontinued in all 4 patients within 8 months but two developed recurrent PAH and were treated satisfactorily with oral therapy. Another group, 40 published its experience treating with epoprostenol 16 patients with moderate and severe POPH. The mPAP decreased to <35 mmhg in 12 75 patients after an average of 7 4 6 8 months treatment and all were listed for lt eleven them underwent without poph related complications the excluded patient had metastasic endometrial cancer epoprostenol was discontinued a median 9 following survival this group similar to transplanted ph it is recommended reassess pah therapy every 3 39,40
Perioperative management of these patients is challenging, and an experienced team should take care of these patients. Active interventions to manage pulmonary pressure and Rv dysfunction during the procedure include the use of epoprostenol or NO as well as inotropic medication such as dobutamine or milrinone. Pulmonary artery catheter and transesophagic echocardiogram are useful monitoring during the procedure. The most critical moments in LT are the induction of anesthesia, during and after graft reperfusion and the immediate postoperative period. After liver reperfusion, there is a massive shift of fluid and release of chemical mediators that can increase pulmonary artery pressure. with these changes, the Rv can fail and as a consequence have hepatic congestion leading to graft failure. The Lv may also fail as a consequence of interventricular dependence mechanism.59,60
Combined Liver Lung Transplant (CLLT) and Combined Heart Lung and Liver Transplant (CHLLT)
To date there are 39 cases reported of combined organ transplant in PAH. Ten of them had POPH, of which 6 underwent CLLT and 4 underwent CHLLT.61 It deserves to be mentioned that 5 patients had no PAH specific therapy before combined transplant.62 with appropriate treatment, these patients may have been LT candidates. One year survival rates after combined transplant are between 70% to 79%, but the value of this information is limited due to the small number of cases.63 Any perceived or reported benefit of combined transplant needs to be weighed against ethical and practical concerns over organ availability and prospect of multiple recipients.
Prognosis and survival
A referral center in France reported survival rates in a pooled population of POPH (n = 154). Survival rates at 1, 3 and 5 years were 88%, 75% and 68%. They found that severity of cirrhosis and CI were major independent prognostic factors.64 A mayor referral center,65 reported its 14 year experience in patients with POPH. Nineteen patients who were not treated with pulmonary vasodilators or LT had a five-year survival of 14%, 54% died in the first year of diagnosis. The five year survival of 43 patients who were treated for POPH but did not undergo LT was 45%, of wich 12% died within the first year of diagnosis. Five year survival in 12 patients who underwent LT was 65% in those who were pretreated with prostanoids vs. 25% in those who were not. They did not find any correlation for mortality with baseline hemodynamic variables, type of liver disease or severity of hepatic dysfunction. More recent data from the REvEAL registry emphasized the gravity of POPH and survival compared to other forms of PAH.66 POPH was associated with the highest hazard ratio (3.6; 95% CI, 2.4 to 5.4) than other causes of PAH.
Conclusions
POPH is a serious complication of portal hypertension, with a significant impact in LT outcomes. For this reason all patients going to LT should be comprehensively evaluated for this condition. Unfortunately all the existing data about medical treatment is based on low evidence studies. LT has improved survival in this population when the patients have been carefully selected and optimally treated.
Correspondencia: Dr. Juan Francisco Moreno Hoyos.
Servicio de Neumología y Cuidados Intensivos. Hospital Universitario Dr. José E. González y Facultad de Medicina U.A.N.L. Av. Madero y Gonzalitos s/n Col. Mitras. Centro Monterrey N.L. C.P. 64460
Correo electrónico: jfmorenoha@gmail.com
Recibido: Julio 2011.
Aceptado: Agosto 2011