


Mauriac syndrome (MS) is a rare complication of type 1 diabetes mellitus (DM1). It is related to low insulin concentrations and is less common since longer-acting insulins became available. It is characterized by hepatomegaly, growth and puberty delay, and the presence of elevated transaminases and serum lipids. The aim of this study was to describe the patients from a pediatric diabetic population that fulfill the criteria of MS.
Materials and methodsA retrospective analysis of the pediatric diabetic population with diagnostic criteria of MS currently followed at Hospital de Braga, was performed.
ResultsFrom a population of 91 patients with DM1 18 years, 6 patients with the criteria for MS were identified: 5 girls, and 1 boy. The age at presentation was 13–17 years, with a minimum interval between DM1 diagnosis and MS criteria of 4 years. All the patients were prescribed intensive insulin therapy (median daily insulin dose: 0.88U/kg). All had a previous history of poor glycemic control before the diagnosis of MS with glycated hemoglobin (HbA1c) between 8.8 and 12.9%. Increase of hepatic enzymes was present in all the patients; 4 of them had associated hepatomegaly. All the girls presented puberty delay and cushingoid features. None of the patients presented short stature and 5 of them presented mixed dyslipidemia.
ConclusionsAlthough MS is an ancient entity described in DM1, it still exists, particularly in adolescent females. Being aware of MS is of extreme importance since most of the clinical features are reversible with better glycemic control.
El síndrome de Mauriac (SM) es una complicación rara de la diabetes mellitus de tipo 1 (DM1), relacionada con bajas concentraciones de insulina, y es menos común desde que están disponibles insulinas de larga duración de acción. Se caracteriza por la hepatomegalia, el retraso del crecimiento y de la pubertad y la elevación de las transaminasas y de los lípidos séricos. El objetivo de este estudio fue la descripción de los pacientes con criterios de SM en una población pediátrica con DM1.
Material y métodosAnálisis retrospectivo de una población pediátrica diabética atendida en el Hospital de Braga con criterios diagnósticos de SM.
ResultadosDe una población de 91 pacientes con DM1 menores de 18 años fueron identificados 6 pacientes con criterios de SM: 5 mujeres y un varón. La edad de presentación fue de 13 a 17 años con un intervalo mínimo entre el diagnóstico de DM1 y SM de 4 años. Todos los pacientes tenían prescrita una terapia insulínica intensiva (dosis media diaria: 0,88U/kg). Todos tenían una historia previa de mal control glucémico antes del diagnóstico de SM con hemoglobina glucosilada (HbA1c) entre 8,8 y 12,9%. Las enzimas hepáticas estaban aumentadas en todos los pacientes y 4 presentaban una hepatomegalia asociada. Todas las niñas mostraban retraso de la pubertad y un aspecto cushingoide. Ningún paciente tenia talla baja y 5 tenían dislipidemia mixta.
ConclusionesAunque el SM es una entidad poco descrita actualmente, aún existe, especialmente en adolescentes femeninas. Identificar el SM es muy importante ya que la mayoría de las alteraciones clínicas son reversibles con mejor control glucémico.
Mauriac syndrome (MS) is a rare complication of type 1 diabetes mellitus (DM1), characterized by hepatomegaly (hepatic glycogenosis), puberty and growth delay, dyslipidemia, transaminase elevation and reduction of IGF1 (insulin-like growth factor 1).1 Cushingoid features may also be present.2
There may be different forms and etiologies involved in Mauriac syndrome. However, there are common features noted in these patients and with adequate insulin treatment there is reversal of growth failure and hepatomegaly if present.3
MS is more common in children and adolescents with poor glycemic control and increased susceptibility of complications,1 and is the commonest cause of hepatic dysfunction in children and adolescents with DM1.4
Although its real incidence is not well-known, due to the reduced number of reported cases in the literature, equal incidence is reported in males and females, with most of the cases occurring in adolescents and young adults.2,5
The authors present a retrospective analysis of patients presenting diagnostic criteria of MS from the type 1 diabetic population followed currently by pediatric endocrinology in Braga's Hospital.
Materials and methodsCases were identified by searching the Pediatric endocrinology DM1 database for confirmed clinical and biochemical features of MS in patients under 18 years at presentation. A retrospective analysis was undertaken. A review of records was conducted to determine patients’ age at presentation of DM1 and MS, as well as glycated hemoglobin (HbA1c) value, hepatic enzymes, lipid profile and median daily insulin dose at MS presentation and follow-up data after MS diagnosis. Data regarding other complications of DM1 (microvascular complications, specifically, retinopathy and nephropathy; macrovascular complications and neuropathies) were also collected. Data research ended in October 30, 2011. The study was approved by the Braga's Hospital pediatric endocrinology coordinator and an informed consent was signed by parents or children legal guardians.
ResultsFrom a population of 91 pediatric patients with DM1 (followed from January 2005 to October 2011), we identified 6 patients with diagnostic criteria for MS: 5 girls and 1 boy.
Regarding the six patients with MS, the median age at DM1 diagnosis was 7.8 years (3–11 years) and at MS presentation 15.3 years (13–17 years). The median interval between DM1 and MS diagnosis was 7.5 years (range 4–14 years). All the patients were under intensive insulin therapy with a median daily insulin dose of 0.88U/kg.
All had a previous history of poor glycemic control prior to the MS diagnosis with a median (glycosylated hemoglobin) HbA1c concentration at diagnosis of 12.3% (range 10.5–13.5%). The median HbA1c of the DM1 population was 8.1% (normal HbA1c<6.5%). Non-compliance regarding nutritional and insulin therapy and self-monitoring was a common feature of all identified MS cases.
At presentation, the 5 girls presented cushingoid features, puberty dysfunction (primary amenorrhea in 2 patients and secondary amenorrhea in 3) and mixed dyslipidemia (high total cholesterol, low HDL[high density lipoprotein] cholesterol and high triglycerides: cut-off values of >200mg/dl for total cholesterol, <40mg/dl for HDL and >180mg/dl for triglycerides). The male patient presented puberty delay and dyslipidemia.
Of the 6 patients, 5 presented hepatomegaly (4 confirmed by abdominal ultrasound), and all showed transaminase elevation: aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels 3–22-fold above the upper normal limit. Liver function tests such as alkaline phosphatase, prothrombin/partial prothrombin time and total bilirubin were normal, as well as thyroid function, IgA and transglutaminase or anti-endomysium levels. None of the patients presented criteria of short stature (all were within the percentiles P10–P75 of expected height for age and sex, according to CDC 2010 Growth Charts).
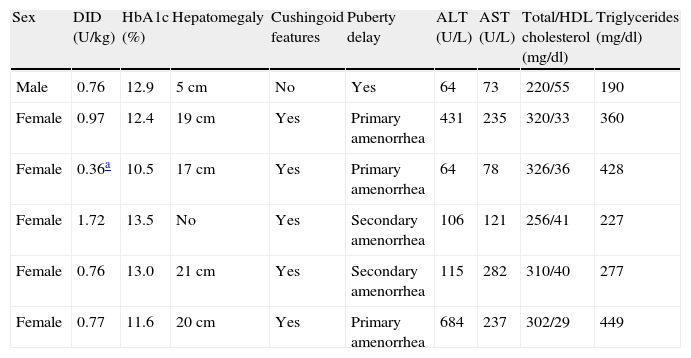
Clinical and biochemical features at MS presentation are shown in Table 1.
Clinical and biochemical features of patients with Mauriac syndrome at diagnosis.
| Sex | DID (U/kg) | HbA1c (%) | Hepatomegaly | Cushingoid features | Puberty delay | ALT (U/L) | AST (U/L) | Total/HDL cholesterol (mg/dl) | Triglycerides (mg/dl) |
| Male | 0.76 | 12.9 | 5cm | No | Yes | 64 | 73 | 220/55 | 190 |
| Female | 0.97 | 12.4 | 19cm | Yes | Primary amenorrhea | 431 | 235 | 320/33 | 360 |
| Female | 0.36a | 10.5 | 17cm | Yes | Primary amenorrhea | 64 | 78 | 326/36 | 428 |
| Female | 1.72 | 13.5 | No | Yes | Secondary amenorrhea | 106 | 121 | 256/41 | 227 |
| Female | 0.76 | 13.0 | 21cm | Yes | Secondary amenorrhea | 115 | 282 | 310/40 | 277 |
| Female | 0.77 | 11.6 | 20cm | Yes | Primary amenorrhea | 684 | 237 | 302/29 | 449 |
DID, daily insulin dose; HbA1c, glycated hemoglobin; ALT, alanine aminotransferase; AST, aspartate aminotransferase; HDL, high-density lipoprotein.
After diagnosis, a stronger multidisciplinary approach (involving endocrinology, pediatrics, nutrition, psychology and psychiatry) was attempted in order to improve glycemic control. The mean duration of follow-up was 1.8 years (1–3.5 years).
A better control was achieved in 2 girls, with a reduction of HbA1c to concentrations below 9% and an improvement regarding puberty impairment (normalization of secondary amenorrhea). Normalization of transaminase concentrations was achieved only in one. A third patient presented transient improved control with normalization of menses and transaminases. In another patient, a normalization of transaminase concentrations was observed with a modest decrease of HbA1c concentration (from 14.5% to 13%). Normalization of transaminase concentrations occurred between 4 and 12 weeks (median 9 weeks) after MS diagnosis. In the sixth patient no better glycemic control was achieved. None of the patients showed an improvement of lipid profile.
The 6 patients had regular ophthalmologic evaluations and none showed signs of diabetic retinopathy. Three of the 6 presented with microalbuminuria (>30mg/dl in 24h).
DiscussionAdolescence is a critical period of development characterized by changes in interpersonal roles, responsibilities and identity construction. This period is more complex for adolescents diagnosed with type 1 diabetes: in addition to experiencing the same challenges as their peers, these adolescents must deal with intensive medical regimens, regular clinic appointments, carbohydrate calculations and frequent daily monitoring of blood glucose levels. Although new insulins and carbohydrate counting approach are currently available to facilitate more optimal glycemic control, many adolescents with type 1 diabetes achieve suboptimal glycemic control and in some cases we can still find MS.
The features of MS are mostly related with deficient insulinization. Patients can develop hepatomegaly due to intrahepatic glycogen deposition; if these patients also have elevated liver enzymes, dyslipidemia, cushingoid features and delayed growth or sexual maturation, Mauriac syndrome can be diagnosed.4 The literature documents decreased blood glucose monitoring frequency and deterioration in blood glucose control and HbA1c, with few adolescents achieving optimal HbA1c values (<7.5%).6 Although being an adolescent greater risk, MS can occur in any age if good metabolic control is not achieved.2
In poorly controlled DM1 patients, the hyperglycemic periods followed by occasional hyperinsulinization and the high cortisol levels as hypoglycemia contra-regulatory hormone lead to hepatic glycogen storage. In hyperglycemic situations, glucose goes freely into the hepatocyte and is also stored as glycogen. On the other hand, the deficient insulinization due to poor glycemic control leads to lipolysis and ketone liberation. Ketosis activates cortisol synthesis promoting the release of fatty acids and hyperglycemia.7,8 A common finding in these patients is the growth delay and/or hypogonadism secondary to the high cortisol levels.4
The fact of the short stature classically described in MS not being reported here might have two explanations: the first is that all the patients were 13–17 years old by the time of diagnosis, meaning that they had already achieved a normal stature before the detioration of metabolic control. The other reason is that data of parents’ height was not available in order to evaluate the expected genetic height, so that some might be in a normal percentile for sex and age but below their genetic potential.
As in other series of cases recently described, also with a small number of patients, the incidence of MS seems to be greater in adolescent girls. The poor glycemic control prior to the diagnosis and the elevated daily insulin dose are also described in other series.4,9 These data suggest the importance of high vigilance in promoting patient compliance to insulin dosing rather than simply increasing insulin dosage in response to hyperglycemia with subsequent weight gain.
With adequate insulin treatment there is a reversal of those features; however, overly aggressive insulin delivery could result in rapid deterioration of diabetic retinopathy and nephropathy.3 The reduction of hepatic enzymes after achieving reasonable glycemic control suggests that liver biopsy and other extensive work-up may be unnecessary in the management of similar patients.9
The authors present this series to stress the importance of high index of suspicion of MS in diabetic adolescents with hepatomegaly, transaminase elevation, dyslipidemia, growth or sexual delay and cushingoid features. A prompt diagnosis and a multidisciplinary approach are the basis of the treatment, but prevention is desirable. Good metabolic control allows improving liver function tests as well as a reduction of the probability of developing other diabetic complications related to persistent hyperglycemia. As this series shows, adolescents and especially females are more vulnerable to this disease and, due to their bio-psycho-social profile, therapeutic adhesion is not easy to achieve, despite new insulin treatments and dietary approach.
Conflict of interestThe authors have no conflict of interest to declare.
Please cite this article as: Dias Joana, et al. El síndrome de Mauriac todavía existe. Endocrinol Nutr. 2012. http://dx.doi.org/10.1016/j.endonu.2012.12.005.
