









In the present work, the effect of varying the carbonate to phosphate (CO32−/PO43−) molar ratios from 0.5 to 4 on the sintering behaviour of carbonated hydroxyapatite (CHA) synthesized by a wet chemical method were investigated. The sintering was performed under carbon dioxide atmosphere at 900°C to maintain the B-type CHA structure. The derived powders as well as the sintered samples were characterized to determine the phase present, crystallographic parameters, the functional group resulting from the substitution of CO32− for PO43−, microstructural evolution under different molar ratios, bulk density, Vickers hardness and fracture toughness. It was found that as the CO32−/PO43− ratio increase, this was accompanied by an increased in the c/a lattice ratio. The sintering studies indicated that the all the CHA was thermally stable and retained the apatite structure after sintering. The relative density of the sintered CHA was found to decrease along with the Vickers hardness and fracture toughness as the CO32−/PO43− ratio increased from 0.5 to 4. The improvement in the mechanical properties was associated with improvement in the relative density and the larger grain size of the sintered samples.
En el presente trabajo, se investigó el efecto de variar las relaciones molares de carbonato a fosfato (CO32-/PO43-) de 0,5 a 4 sobre el comportamiento de sinterización de la hidroxiapatita carbonatada (CHA) sintetizada por un método químico húmedo. La sinterización se realizó bajo una atmósfera de dióxido de carbono a 900° C para mantener la estructura de CHA de tipo B. Los polvos derivados y las muestras sinterizadas se caracterizaron para determinar la fase presente, los parámetros cristalográficos, el grupo funcional resultante de la sustitución de CO32 por PO43, la evolución microestructural en diferentes relaciones molares, la densidad aparente, la dureza Vickers y la resistencia a la fractura. Se encontró que a medida que aumentaba la relación CO32−/PO43−, esto iba acompañado de un aumento en la relación de celosía c/a. Los estudios de sinterización indicaron que todo el CHA era térmicamente estable y retuvo la estructura de apatita después de la sinterización. Se encontró que la densidad relativa del CHA sinterizado disminuía junto con la dureza Vickers y la resistencia a la fractura, ya que la relación CO32−/PO43− aumentó de 0.5 a 4. La mejora en las propiedades mecánicas se asoció con una mejora en la densidad relativa y la mayor Tamaño de grano de las muestras sinterizadas.
The major component of bone is a composite of inorganic compound (carbonated HA) and organic compound (collagen) [1]. However, bones are also prone to injury and defects resulting from many different causes. Hence, it is logical to say that large bony defect due to injury or disease can significantly alter one's body equilibrium and quality of life [2]. Thus, bone grafts are required to reconstruct damaged hard tissues or to treat poor bone-healing conditions. Bone grafts are materials of natural or synthetic origin and in general can be classified as autograft (harvested from the patient itself), allograft (taken from a donor's body), xenograft (taken from animal such as bovine) and synthetic bone graft fabricated from synthetic biomaterials to mimic natural bone tissue [2,3].
Amongst them, synthetic bone graft is the preferred choice as it circumvents many of the disadvantages faced using bone grafts obtained from donors [3–5]. Therefore, various kinds of biomaterials have been developed as bone substitute such as bioceramics, metals, polymers and composites. Among these materials, the family group of calcium phosphate, especially hydroxyapatite (HA) has been used extensively as bone graft substitute due to its excellent biocompatibility, bioactivity and nontoxicity properties [6–16]. In comparison to HA, biological apatites are non-stoichiometric, poorly crystalline, and contain several foreign ions, mainly carbonate CO32− (2–8wt%) [17–20]. Carbonated hydroxyapatite has been found to be easily resorbed by living cells and to possess higher solubility than stoichiometric HA which is free of carbonate ions [21,22]. Furthermore, substitution of CO32− ions into the HA lattice has been reported to enhance the mechanical properties of the bioceramic [23]. Depending on the mode of substitution, CHA can either be classified as A-type CHA (CO32− substitutes OH−), B-type CHA (CO32− substitutes PO43−) or AB-type CHA (CO32− substitutes both OH− and PO43−) [23–25]. Biological apatites are predominantly B-type CHA with A/B-type ratio of 0.7–0.9. Higher A/B-type ratio was observed in old tissue as compared to the young one [23,26]. Various methods have been proposed to synthesis nano-crystalline B-type CHA. However, in principle there are two main methods used to synthesize CHA powder, namely wet and dry method. The wet chemical method is considered as the most common synthesis method to produce B-type CHA powder [27–34].
Some researchers evaluated the effects of carbonate content on physical and bioactivity properties of CHA [24–31]. However, the influence of varying the CO32−/PO43− ratio on the sintering behaviour of synthesized B-type CHA has not been systematically studied. Therefore, the aim of this research is to evaluate the effect of varying the CO32−/PO43− ratio on the physical and mechanical properties of B-type CHA prepared via a wet chemical method and sintered at 900°C under CO2 atmosphere.
ExperimentalIn the present work, a wet chemical method was used to produce B-type CHA [34] by using ≥ 99% purity starting materials comprising of di-ammonium hydrogen phosphate (NH4)2HPO4, ammonium bicarbonate NH4HCO3 and calcium nitrate tetrahydrate Ca(NO3)2.4H2O as the precursors for PO43−, CO32− and Ca2+, respectively. During the synthesis process, the calcium to phosphate ratio was maintained at 1.67 for all variant substitutions. Five different variation of CO32−/PO43− molar ratios, i.e. 0.5 to 4, were prepared and the as-synthesized CHA powders were labelled as 0.5CHA, 1CHA, 2CHA, 3CHA and 4CHA, respectively. A stoichiometric HA was also synthesized under similar condition for comparison purpose.
In a typical synthesis process, the procedure involves preparing an aqueous solution of NH4HCO3 and (NH4)2HPO4, respectively in 60ml of deionized water. Both transparent aqueous solutions were subsequently mixed together for 15min at a stirring rate of 450rpm. Next, 1M NaOH solution was added dropwise into the mixture of NH4HCO3 and (NH4)2HPO4 solution to adjust the pH to 11. Another solution was prepared by dissolving a specific amount of Ca(NO3)2.4H2O in 100ml of acetone. Subsequently, the organic solution was poured directly into the previously stirred aqueous solution at a rate of 50ml per sec. resulting in the formation of a slightly milky solution. After that, the precipitate was filtered and washed with 1500ml of deionized water. The white cake obtained was dried in a box oven at 100°C for 20h and subsequently fine powder was obtained by sieving through a 100μm sieve [34].
The prepared powders were compacted to produce disc (20mm in diameter) samples and then subjected to cold-isostatic pressing at 200MPa. The samples were then sintered in a CO2 atmosphere (flow rate of 4ml/min) at a temperature of 900°C for 2h, at a cooling and heating rate of 10°C/min. This temperature was chosen in order to ensure that the CHA produced is of the B-type since it is well established that sintering above 900°C would cause an increase in the a-axis parameter which is typical for an A-type substitution and may also result in the decomposition of the CHA phase [33].
The phases present in the powder and sintered samples were determine by X-ray diffraction (XRD) (PANalytical X’Pert3 Powder Diffractometer) from 2θ=20–50°, at a scan speed of 4°/min and a step of 0.02° using CuKα radiation (40kV, 40mA). Phase determinations were made using Standard International Centre for Diffraction Data (ICDD) card no. 9-0432 for HA. Determination of the lattice constants of the samples was obtained by Rietveld refinement of the XRD data which is processed by Fullprof program according to the procedure described in [35]. In general, the peak fittings were performed using Pseudo-Voigt profile distribution and the Marquardt least-squares procedures were used to minimize the difference between the observed and simulated diffraction patterns. The various reliability index parameters (R) and goodness of fit (χ2) were obtained. Subsequently, the goodness of fit indicator (GoF) is calculated by dividing the numerical values obtained for the residuals weighted pattern (Rwp) with the expected error (Rexp) to ascertain the quality of fit between the observed and simulated diffraction patterns [35]. The refinements were initially based on the literature data [36] using the hexagonal crystal (space group P63/m) for HA with lattice parameters a=b=9.4219Å and c=6.8822Å.
The types of functional groups present in the samples were determined by using Fourier transform infrared (FTIR) spectrometry (Perkin Elmer spectrometer, USA) at wavelength interval from 400cm−1 to 4000cm−1. X-Ray Fluorescence (XRF) (Rigaku Rix 3000, Japan) was used to determine the Ca/P ratio of the CHA samples. The amount of CO32− present in the powders was calculated from the carbon content [25] measured using the CHNS/O elemental analyzer (Perkin Elmer, USA). The microstructural evolution of the sintered samples at different CO32−/PO43− ratios were examined on fracture surface using a Carl Zeiss Auriga field emission scanning electron microscopy (FESEM). The average grain size of the sample was determined from the line intercept analysis [37]. The weight loss of the samples during heat treatment was studied from room temperature to 1000°C by using a thermo-gravimetric analyser (Perkin Elmer Pyris TGA-6). The density of the sintered samples was determined by Archimedes’ method using distilled water. The linear shrinkage of the samples after sintering was also determined from the change in diameter.
The Vickers indentation method was used to determine the Vickers hardness (Hv) and fracture toughness (KIc) of sintered ceramics. For this test, the sample surface has to be reflective in order to visualize the indent diagonals and cracks. Therefore the samples were ground successively using silicon carbide papers with grit sizes varying from 600 (rough) to 1200 (fine) and followed by mirror-polish to 1μm surface finish using diamond paste. The indentation was made on the surface using a pyramidal diamond indenter (Shimadzu HMV) with an applied load of 1.96N. During the test, the load was applied gradually and was held in position for 10s before taking the measurements. The fracture toughness was determined by using the equation proposed by Niihara [38] as shown in Eq. (1). For each composition, five samples were tested and three indents were made for each sample.
where a is the half diagonal of the indentation, c is the radial crack dimension measured from the centre of the indent impression, i.e. c=L+a and L is the crack length generated by indentation.Results and discussion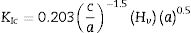
The XRD signatures of the synthesized HA and CHA powders are as shown in Fig. 1. The diffraction peaks of the synthesized HA and 0.5CHA to 4CHA powders exhibited a single phase material that closely match the ICDD standard for hydroxyapatite. Further increased in the CO32−/PO43− ratio above 4, however resulted in the development of calcium carbonate phase in the powder (not shown here). This excess amount of CO32− ions present during synthesis could have reacted with Ca2+ in the apatite structure to form calcium carbonate due to the limited PO43− available for the substitution [24,28,39,40].

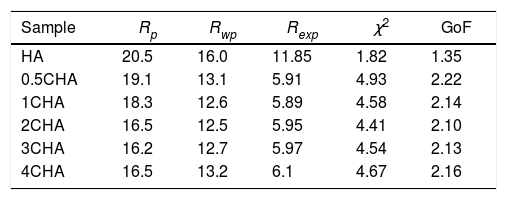
The Rietveld refinement of the sintered samples showing the observed (IO) and calculated (IC) patterns are presented in Fig. 2. A very small differences could be seen between both patterns as shown by the difference curve (IO−IC) plotted under the respective XRD signatures. There was no secondary phase formation detected and all the samples conformed to the standard ICDD for HA phase. The various R factors of these samples are presented in Table 1. In general, the GoF values for the CHAs were found to be slightly higher (i.e. 2.10–2.22) if compared to the control HA which had a lower value of 1.35 i.e. demonstrating a very good fit exist between the observed and calculated patterns. This is expected since the CHA are non-stoichiometric apatite and their calcium to phosphate proportion was found to increase with increasing CO32−/PO43− ratio, from 1.86 for 0.5CHA to 1.96 for 4CHA. Nevertheless, Table 1 shows that for all samples, both the IO and IC patterns are in good agreement with each other and this is considered acceptable in accordance to the basic rule of GoF less than 4% [35,41,42].
 and calculated (IC) patterns.")
The various reliability index parameters obtained from the Rietveld refinement for the sintered samples.
| Sample | Rp | Rwp | Rexp | χ2 | GoF |
|---|---|---|---|---|---|
| HA | 20.5 | 16.0 | 11.85 | 1.82 | 1.35 |
| 0.5CHA | 19.1 | 13.1 | 5.91 | 4.93 | 2.22 |
| 1CHA | 18.3 | 12.6 | 5.89 | 4.58 | 2.14 |
| 2CHA | 16.5 | 12.5 | 5.95 | 4.41 | 2.10 |
| 3CHA | 16.2 | 12.7 | 5.97 | 4.54 | 2.13 |
| 4CHA | 16.5 | 13.2 | 6.1 | 4.67 | 2.16 |
Note: χ2=(Rwp/Rexp)2.
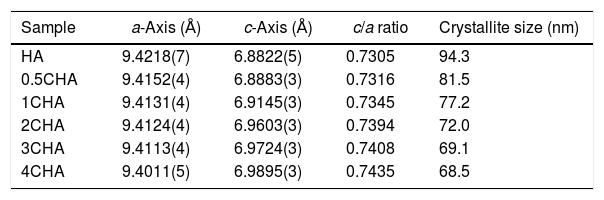
The structural parameters obtained from the Rietveld refinement of all the samples have been determined based on the unit cell parameters at the hexagonal setting (space group P63/m) as given in Table 2 which further verifies the formation of apatite structure. The results show that in comparison to the synthesized HA, the c/a parameter increases with increasing CO32−/PO43− ratio resulting from a decrease in the a-axis and an increase in the c-axis, which could be attributed to the replacement of the bigger tetragonal PO43− by smaller planner CO32− ion on the B-sites [17]. The carbonate substitution would induce lattice defects in the apatite structure and suppress crystal growth [24,28,39,40,43–51]. This is evident from the decreased in the crystallite size (Table 2) and the decreased in the crystallinity of the powders with increasing CO32−/PO43− ratio as depicted by the XRD peaks (Fig. 1) which declined in both sharpness and intensity. In addition, the typical features of the (211) and (112) XRD peaks at 2θ about 32° observed for stoichiometric HA as shown in Fig. 1 becomes unnoticeable for all the CHA samples due to the peaks overlapping each other.
Structural parameters as determined for the sintered samples from the Rietveld refinement.
| Sample | a-Axis (Å) | c-Axis (Å) | c/a ratio | Crystallite size (nm) |
|---|---|---|---|---|
| HA | 9.4218(7) | 6.8822(5) | 0.7305 | 94.3 |
| 0.5CHA | 9.4152(4) | 6.8883(3) | 0.7316 | 81.5 |
| 1CHA | 9.4131(4) | 6.9145(3) | 0.7345 | 77.2 |
| 2CHA | 9.4124(4) | 6.9603(3) | 0.7394 | 72.0 |
| 3CHA | 9.4113(4) | 6.9724(3) | 0.7408 | 69.1 |
| 4CHA | 9.4011(5) | 6.9895(3) | 0.7435 | 68.5 |
A typical thermal analysis of the sample 4CHA is presented in Fig. 3. In general, the TG curve shows two stage of significant weight loss occurring during heating. From room temperature to 500°C (stage I), there was about 12wt% loss which can be ascribed to the liberation of absorbed and lattice H2O [33,40]. This was followed by another weight loss of about 3.5wt% from 500 to 900°C (stage II) and this could be associated with decomposition of carbonate. There was no further weight loss observed from 900 to 1000°C. A similar trend was observed for all other samples and the total weight loss measured at 900°C for the samples did not vary very much as shown in the inset table in Fig. 3. In general, the amount of carbonate loss during heating of the synthesized powders is considered below the threshold limit for the stabilization of the B-type CHA. It is also believed that the sintering of the samples in CO2 atmosphere could have compensated the loss of CO32− since the characteristics of the CHA phase were preserved as confirmed by the XRD analysis.
 subjected to heating from room temperature to 1000°C. The inset table shows the total weight loss measured for the other samples.")
The FTIR spectra of the sintered CHA with variant carbonate content are shown in Fig. 4. It was found that the intensity of the CO32− derived bands in the FTIR spectra increased proportionally with increasing CO32−/PO43− ratio. The two regions of the spectra that represents the characteristic of B-type CHA are the CO32− vibrations detected at 871–873cm−1 (ν2) and 1416–1486cm−1 (ν3) [24,25]. The synthesized HA has a sharp peak at approximately 3570cm−1 that corresponds to the stretching vibration of the lattice OH− ions and the relatively broad peak centred around 3432cm−1 and the minor peak at 1642cm−1 are attributable to absorbed water which is a typical feature observed for stoichiometric HA [27,48,49]. The characteristic bands of PO43− groups of the apatite structure were observed at about 566–568 and 603–604cm−1 (ν4) and 1039–1046cm−1 (ν3) for all samples. The significant changes observed in the spectra of the CHA are the absences of the broad peaks associated with absorbed water and that the OH− peak at 3570cm−1 appears to be of a lower intensity compared to the HA sample, suggesting less OH groups in the carbonated-substituted HA. This is expected due to the non-stoichiometric nature of the CHA. In addition, the absences of CaCO3 at 712cm−1 in all spectra confirmed that the HA phase was not disrupted after sintering and this is in good agreement with the XRD analysis. The amount of carbonate content present in the sintered samples for 0.5CHA, 1CHA, 2CHA, 3CHA and 4CHA as per the CHNS/O analysis were 2.01, 2.95, 4.10, 5.05 and 5.25wt%, respectively. These values are within the limit of 2–8wt% normally found in human bones [47].

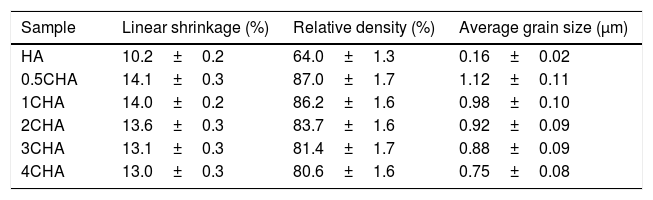
The relative density and linear shrinkage of the sintered samples are given in Table 3. A relative density of more than 80% was obtained for the CHA samples and this is in good agreement with that reported by other researchers [33]. The highest relative density of 87% was measured for the 0.5CHA sample and this value decreases slightly with increasing carbonate content. The linear shrinkage correlated well with the relative density and the values were found to be high, at about 13–14%. In addition, the carbonate substitution was found to be beneficial in enhancing densification when compared to the unsubstituted HA which exhibited a relatively low density of about 64% which correspond to a low shrinkage of about 10% when sintered under similar conditions. The higher porosity of the HA can be observed from the FESEM of the fractured surface micrograph shown in Fig. 5. In general, all the samples exhibited an equiaxed and uniform grain morphology. The synthesized HA exhibited a fine microstructure, composed of almost spherical grains, having an average size of about 0.16±0.02μm. From the fracture morphology, the HA revealed a smooth fracture associated with an intergranular-type fracture whereas a transgranular-type fracture is observed for the CHA samples.
Relative density, linear shrinkage and grain size of the sintered samples.
| Sample | Linear shrinkage (%) | Relative density (%) | Average grain size (μm) |
|---|---|---|---|
| HA | 10.2±0.2 | 64.0±1.3 | 0.16±0.02 |
| 0.5CHA | 14.1±0.3 | 87.0±1.7 | 1.12±0.11 |
| 1CHA | 14.0±0.2 | 86.2±1.6 | 0.98±0.10 |
| 2CHA | 13.6±0.3 | 83.7±1.6 | 0.92±0.09 |
| 3CHA | 13.1±0.3 | 81.4±1.7 | 0.88±0.09 |
| 4CHA | 13.0±0.3 | 80.6±1.6 | 0.75±0.08 |
 HA, (b) 0.5CHA, (c) 1CHA, (d) 2CHA, (e) 3CHA and (f) 4CHA.")
In agreement with the higher relative density and higher shrinkage values, a much compact structure, comprised of large grains were observed for all the CHA samples. The measured grain size of the sintered CHA was found to decrease monotonically with increasing carbonate content, from an average value of 1.12±0.11μm to 0.75±0.08μm (see Table 3). This decreased in the grain size, however was found to have effect on the mechanical properties of the samples as depicted in Fig. 6.

As can observed in Fig. 6, the general trend is that the mechanical properties decreases with increasing carbonate content. In agreement with the density results, the Vickers hardness of the CHA was also higher (>1.4GPa) when compared to 1.04GPa obtained for HA when sintered at 900°C and 1.17GPa reported for HA sintered at 1000°C [52]. The improvement in the mechanical properties of the CHA could be associated with the improvement in the bulk density of the sintered compact as well as in part to the increased in the c/a lattice parameter ratio as depicted in Table 2. It is envisaged that the distortion of the a-axis in the CHA lattice would create additional dislocation barrier for plastic deformation to proceed, thus resulting in improvement in the mechanical properties. The present study also revealed that smaller grain size may not necessarily result in better toughness and there is certainly a combinatory effect between density, grain size and carbonated content. More work is in progress to elucidate this behaviour. Other researchers have also reported that carbonate substitution was beneficial in enhancing the mechanical and biological properties compared to hydroxyapatite [53,54] although the exact role of carbonate in enhancing the mechanical properties is not well understood.
ConclusionsIn the present work, the effect of varying the CO32−/PO43− ratio from 0.5 to 4 on the densification behaviour of CHA was investigated. Phase analysis revealed that the phase stability and B-type apatite structure were not disrupted after sintering. Nevertheless, increasing the CO32−/PO43− ratio was found to increase the c/a ratio of the CHA and resulted in a concomitant decreased in the grain size of the sintered samples. In addition, the relative density, Vickers hardness and fracture toughness of the sintered CHA were found to decrease with increasing CO32−/PO43− ratio from 0.5 to 4 with values varying from 87 to 80% of theoretical, 1.97 to 1.36GPa and 1.97 to 1.36MPam1/2, respectively. In comparison to the stoichiometric HA, the carbonate substitution has generally improved the densification and mechanical properties of the sintered body, thus revealing great potential for use as a synthetic bone graft biomaterial.
The authors gratefully acknowledge UM and USM for the technical assistance to carry out this research. This study was supported under the FRGS grant no. FP020-2018A.

