



Haemophagocytic syndrome (HS) is a rare disease that is often fatal despite treatment. HS is characterized by fevers, lymphadenopathy, hepatosplenomegaly, cytopenias and hyperferritinaemia due to deregulated activation and proliferation of macrophages, leading to uncontrolled phagocytosis of platelets, erythrocytes, lymphocytes, and their hematopoietic precursors throughout the reticuloendothelial system. Mycobacterium tuberculosis-associated HS is a rare and underdiagnosed association with only 39 cases reported. We describe a case of HS associated with disseminated Mycobacterium tuberculosis in the setting of post-liver transplantation anti-hepatitis C therapy with pegylated interferon (pegIFN), ribavirin (RBV) and telaprevir (TVR). Despite the delay in the etiologic diagnosis, the patient was treated properly with corticosteroids, cyclosporine and tuberculostatic agents. It is unknown whether telaprevir, a drug that only recently has been started off-label in liver transplant recipients, may have contributed to the development of the HS. Unfortunately, as in many reported cases of HS, the outcome was unfavourable resulting in the death of the patient.
Haemophagocytic syndrome (HS), more properly referred to as haemophagocytic lymphohistiocytosis, is a rare disease that is often fatal despite treatment.1 HS is characterized by fevers, lymphadenopathy, hepatosplenomegaly, cytopenias and hyperferritinaemia due to deregulated activation and proliferation of macrophages, leading to uncontrolled phagocytosis of platelets, erythrocytes, lymphocytes, and their hematopoietic precursors throughout the reticuloendothelial system.2 HS can be either primary, with a genetic etiology, or secondary, associated with malignancies, autoimmune diseases or infections. Epstein-Barr virus is the most common infectious etiology implicated in HS,3 but the syndrome has been associated with a variety of other viral, bacterial, and parasitic pathogens. We describe a case of HS associated with disseminated Mycobacterium tuberculosis in the setting of post-liver transplantation anti-hepatitis C therapy with pegylated interferon (pegIFN), ribavirin (RBV) and telaprevir (TVR).
Case ReportA 63 year old man transplanted 4 years ago for hepatitis C virus (HCV) related cirrhosis, presented with a 5-day history of fever (without chills or sweats) and malaise with diffuse, non-pruriginous rash involving the trunk, lower limbs and axillae.
He had a history of recurrent HCV hepatitis with markers of de novo autoimmunity diagnosed eight months post-transplant. Treatment with azathioprine and prednisone was followed by clinical and analytical improvement with several flares occurring when prednisone was tapered to 5 mg/day. Despite immunosuppressive therapy, a sudden deterioration of liver function tests occurred and, in contrast to previous graft biopsies, no histological markers of auto-immunity were observed and fibrosis was shown to have progressed to F3-4/6 (Ishak score). Treatment with triple therapy (pegIFN, RBV and TVR) was initiated 6 weeks before admission with excellent biochemical and viral responses. Azathioprine and prednisone doses were the same before and during antiviral therapy (azathioprine 75 mg daily and prednisone 5 mg daily). Cyclosporine doses were changed at the start of triple therapy (from 50 mg/12 h before starting triple therapy to 25 mg/day at the start of antiviral therapy and subsequently modified according to trough levels). A couple of weeks post-admission, immunosuppressive doses were increased (azathioprine 75 mg/day, prednisone 10 mg/day and cyclosporine 25 mg/12 h) due to significant impairment of liver function tests and high suspicion of autoimmune component.
On physical examination, the patient was in good condition. He was haemodynamically stable, with fever of 38 degrees and lack of neurological focality. A confluent rash with erythematous macules was distributed on the trunk, legs and armpits. There were no cervical, supraclavicular, axillary or inguinal lymphadenopathies and genitourinary and neurological examinations were unremarkable.
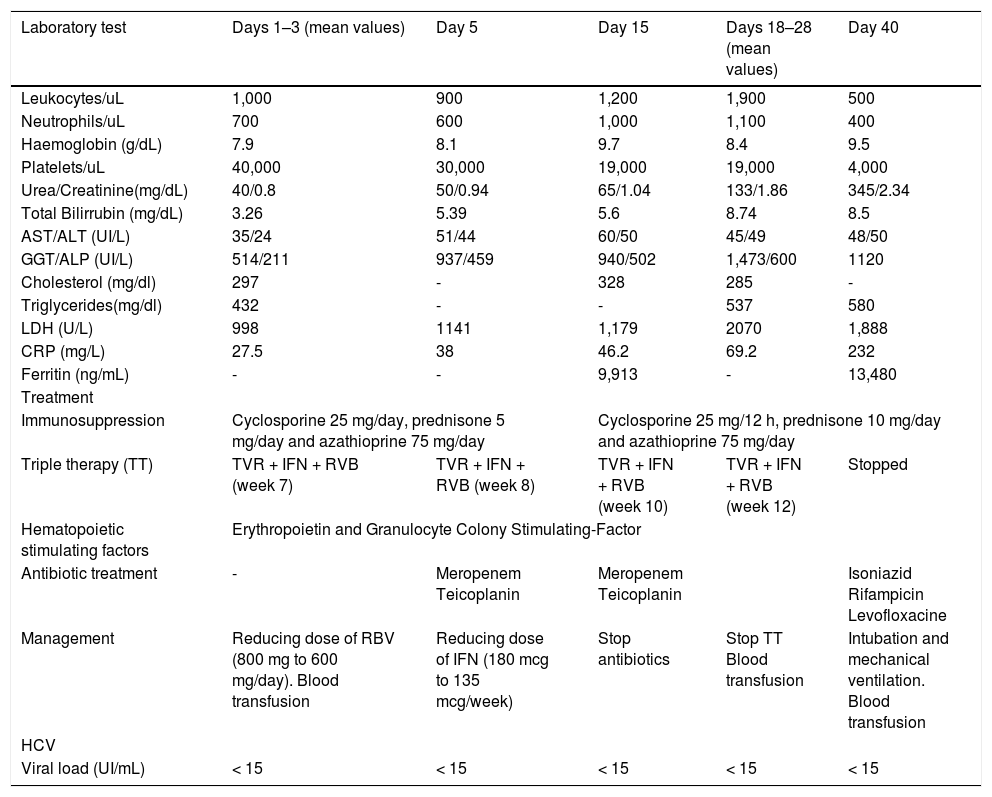
Laboratory evaluation revealed a progression of the already known pancytopenia (leukocytes 1,000/uL; neutrophils 700/u.L; platelets 40,000/u.L, haemoglobin 7.9 g/dL) which was thought to be associated with antiviral treatment; it that had already required blood transfusion twice as well as reduction of the initial RBV dose (800 mg/day initial dose, current dose 600 mg). He also had slight worsening of liver function tests with regards to previous controls [bilirubin 3.26 mg/dL, aspartate aminotransferase (AST)/ alanine aminotransferase (ALT) 35/24 UI/L, gamma-glutamyl transferase (GGT)/alkaline phosphatase (ALP) 514/211 UI/L] and C-reactive protein (CRP) 27.5 mg/dL. HCV viral load was undetectable (< 15 UI/mL) from the fourth week of treatment with triple therapy. Complementary examinations initially requested (urinalysis; urine, blood and sputum cultures; citomegalovirus, Epstein-Barr virus and parvovirus B19 viral loads; tuberculin skin testing; chest radiography and abdominal ultrasound) were normal. Given these findings, empirical treatment with meropenem and teicoplanin was started. In addition, the rash was evaluated by dermatology, and as initially suspected, a diagnosis of skin rash type I associated with TVR was made and topical treatment was undertaken.
Despite empirical antibiotic therapy, the fever persisted with the same characteristics without clinical deterioration or infectious focality. Repetitive microbiological cultures remained negative. In successive analytical controls, a slight deterioration of renal function was observed (urea 44 mg/dL, creatinine 1.18 mg/dL) together with hypercholesterolemia and hypertriglyceridemia (total cholesterol 258 mg/dL, triglycerides 336 mg/dL), impaired liver function tests with a cholestatic pattern (bilirubin 5.39 mg/dL, AST/ALT 51/44 U/L, GGT/ALP 937/ 439 U/L), hyperferritinemia (ferritin 12,461 ng/ mL) and increase of other acute phase reactants (CRP 46.2 mg/dL; GSV 65 mm/h; procalcitonin 3.98 ng/mL; fibrinogen 606 mg/dL). In addition, worsening pancytopenia with normocytic-normochromic anemia (leukocytes 800/µL, neutrophils 700/µL, hemoglobin 7.7, platelets 20,000/µL) occurred. Blood transfusion (three times) and treatment with hematopoietic stimulating factors (erythropoietin and granulocyte colony-stimulating factor) were required, and the dose of pegIFN was reduced to 135 mcg per week. Thyroid hormones and autoimmunity studies were negative. The echocardiogram showed no abnormalities.
A liver biopsy revealed a granulomatous non-necrotizing hepatitis. Polymerase Chain Reaction (PCR) tests to detect mycobacterial DNA and Ziehl-Neelsen of the liver biopsy sample were negative. In addition, the patient had no history that suggested acute exposure to Mycobacterium tuberculosis. Based on these findings, a search of granulomatous diseases was undertaken. The study was completed with serological tests of Leishmania, syphilis, Coxiella, Chlamydia, Brucella and Toxoplasma, all of which were negative. Angiotensin converting enzyme (ACE) showed high levels (76 U/L) with a normal calcium value. The bone marrow aspirate demonstrated a very hypocellular marrow with intense haemophagocytosis signs. The bacterial and fungal cultures of bone marrow were negative. However, Mycobacterium tuberculosis DNA by PCR was detected in bone marrow.
Weeks later, blood and bone marrow cultures grew Mycobacterium tuberculosis on Lowenstein-Jensen medium (at 4 and 3 weeks, respectively). The isolate was sensitive to isoniazid, rifampicin, pyrazi-namide and ethambutol.
Given the constellation of fever, pancytopenia, hyperferritinaemia, hypertriglyceridemia in the presence of haemophagocytosis within the bone marrow, a diagnosis of HS was made, probably associated with disseminated tuberculosis.
Following the discovery of the etiologic agent, tuberculosis treatment was initiated. Meanwhile, supportive treatment continued with hematopoietic stimulating factors and blood components transfusion. HCV treatment was stopped after 3 months of antiviral therapy. Despite these measures, the patient continued to deteriorate clinically. In addition, he developed a transfusion-related acute lung injury (TRALI) requiring intubation and mechanical ventilation. Finally, a pulmonary infection and acute renal failure caused the patient’s death 40 days following the initial admission (Table 1).
Development and management of the patient from the onset of symptoms.
| Laboratory test | Days 1–3 (mean values) | Day 5 | Day 15 | Days 18–28 (mean values) | Day 40 |
|---|---|---|---|---|---|
| Leukocytes/uL | 1,000 | 900 | 1,200 | 1,900 | 500 |
| Neutrophils/uL | 700 | 600 | 1,000 | 1,100 | 400 |
| Haemoglobin (g/dL) | 7.9 | 8.1 | 9.7 | 8.4 | 9.5 |
| Platelets/uL | 40,000 | 30,000 | 19,000 | 19,000 | 4,000 |
| Urea/Creatinine(mg/dL) | 40/0.8 | 50/0.94 | 65/1.04 | 133/1.86 | 345/2.34 |
| Total Bilirrubin (mg/dL) | 3.26 | 5.39 | 5.6 | 8.74 | 8.5 |
| AST/ALT (UI/L) | 35/24 | 51/44 | 60/50 | 45/49 | 48/50 |
| GGT/ALP (UI/L) | 514/211 | 937/459 | 940/502 | 1,473/600 | 1120 |
| Cholesterol (mg/dl) | 297 | - | 328 | 285 | - |
| Triglycerides(mg/dl) | 432 | - | - | 537 | 580 |
| LDH (U/L) | 998 | 1141 | 1,179 | 2070 | 1,888 |
| CRP (mg/L) | 27.5 | 38 | 46.2 | 69.2 | 232 |
| Ferritin (ng/mL) | - | - | 9,913 | - | 13,480 |
| Treatment | |||||
| Immunosuppression | Cyclosporine 25 mg/day, prednisone 5 mg/day and azathioprine 75 mg/day | Cyclosporine 25 mg/12 h, prednisone 10 mg/day and azathioprine 75 mg/day | |||
| Triple therapy (TT) | TVR + IFN + RVB (week 7) | TVR + IFN + RVB (week 8) | TVR + IFN + RVB (week 10) | TVR + IFN + RVB (week 12) | Stopped |
| Hematopoietic stimulating factors | Erythropoietin and Granulocyte Colony Stimulating-Factor | ||||
| Antibiotic treatment | - | Meropenem Teicoplanin | Meropenem Teicoplanin | Isoniazid Rifampicin Levofloxacine | |
| Management | Reducing dose of RBV (800 mg to 600 mg/day). Blood transfusion | Reducing dose of IFN (180 mcg to 135 mcg/week) | Stop antibiotics | Stop TT Blood transfusion | Intubation and mechanical ventilation. Blood transfusion |
| HCV | |||||
| Viral load (UI/mL) | < 15 | < 15 | < 15 | < 15 | < 15 |
ALP: alkaline phosphatase. AST: aspartate aminotransferase. ALT: alanine aminotranferease. CRP: C -reactive protein. GGT: gamma-glutamyl transferase. IFN: interferon. LDH: lactate dehydrogenase. RBV: ribavirin. TT: triple therapy. TVR: telaprevir.
We report a case in which the history of patient autoimmunity, treatment with antiviral triple therapy and its associated side effects (fever, rash and pancytopenia) and the status of immunosuppression, made a differential diagnosis extremely difficult. HS was suspected when the nearly acellular bone marrow with intense hemophagocytosis was observed in a patient with severe pancytopenia. The isolation of Mycobacterium tuberculosis in two different samples (bone marrow and blood) and the detection of its genome, would explain the fever, increased reactants and the development of HS.
Haemophagocytic syndrome, or haemophagocytic lymphohistiocytosis, is a life-threatening clinico- pathological entity characterized by an impaired or absent function of natural killer (NK) cells and cytotoxic T cells. This dysregulation results in uncontrolled and ineffective immune activation leading to cellular damage and multi-organ failure as well as proliferation and activation of benign macrophages with haemophagocytosis throughout the reticuloendothelial system.4 Clinical signs of HS include prolonged fever, lymphadenopathy, hepatosplenomegaly, and a maculopapular rash. Laboratory abnormalities include cytopenias, raised levels of serum ferritin, triglycerides and bilirubin, and low fibrinogen. Poor prognostic factors include age greater than 30 years, hyperferritinaemia, disseminated intravascular coagulation, and increased beta2-microglobulin levels.1,5 HS is divided into primary or genetic HS and secondary or reactive HS. Primary HS presents during infancy and is associated with a high mortality rate. Secondary HS, which this patient developed, may occur at any age, most commonly in the setting of malignancy (particularly T-cell lymphoma), autoimmune diseases, drug hypersensitivity reaction or infection and has a better prognosis.1 EBV is the most common infectious etiology implicated in HS, but the syndrome has been associated with a variety of other viral, bacterial and parasitic pathogens, including HIV, cytomegalo-virus, Salmonella typhi, Histoplasma capsulatum, Leishmania species, Mycobacterium tuberculosis, and Toxoplasma gondii.2,5 Patients have been treated with various immunomodulatory therapies, including dexamethasone, intravenous immunoglobulins, antithymocyte globulins, cyclosporine and etoposide-based chemotherapeutic regimens.
In our patient, the maculopapular rash was initially attributed to TVR. In TVR trials, approximately half of treated patients developed rash. More than 90% of these events were grade 1 or 2 (mild/moderate) and in the majority (92%) of cases, progression to a more severe grade did not occur.6 In addition, adverse events were reported more frequently in the TVR triple therapy arm than in patients treated with dual pegIFN/RBV.7 Known adverse events associated with pegIFN/RBV include psychiatric disorders (depression, anxiety and insomnia) and blood disorders (mainly anemia and neutropenia, and less frequently thrombocytopenia).8,9 In our patient, both the rash and the pancytopenia were initially attributed to antiviral treatment. However, persistent fever and worsening of blood tests required further research.
The finding of incomplete non-caseating granulomas in the liver biopsy led to a search of additional causes of hepatic granulomas, including sarcoidosis, autoimmune or infectious diseases, drugs, or neo-plastic conditions.10,11 In our case, the initial absence of microbiological isolation, the presence of non-caseating granulomas and the elevation of ACE made us think of sarcoidosis. Indeed, sarcoidosis has been described in the setting of IFN therapy and has also been described as a cause of HS;12.–14 it may manifest as a systemic or cutaneous disease or a combination of both with pulmonary and skin involvement being the most frequent sites involved. In our case, the lack of lung involvement was against this diagnosis.
Finally, the presence of Mycobacterium tuberculosis DNA in bone marrow and growth in blood and bone marrow cultures led to the final diagnosis: a HS due to disseminated tuberculosis. There are only 39 published cases of HS associated with tuberculosis, 37 cases of which recently reviewed.2,15,16 Fever was present in all cases and organomegaly in 75%. Extrapulmonary tuberculosis was observed in 83% of patients. Pancytopenia, particularly thrombocytopenia, was common (89% of patients). Twenty-nine patients received therapy, either antituberculous drugs alone (nine patients) or a combination of anti-tuberculous drugs with immunomodulatory therapy (20 patients). Immunomodulatory treatment mostly consisted of steroids but two patients underwent splenectomy and two plasmapheresis. Twelve out of the 20 patients who received a combination of immunomodulatory and antituberculous treatment and seven of nine patients who received antituberculous treatment alone survived. Patients who received no treatment died.1,2 In summary, tuberculosis-associated HS carries a poor prognosis with approximately 50% mortality; but antituberculous and immunomodulatory therapies may improve this prognosis. Therefore, tuberculosis should be considered early in the differential diagnosis of infectious etiologies associated with HS.2,17 A negative tuberculin skin test should never preclude the consideration of tuberculosis, and confirmation of the diagnosis is often made from microscopical examination of infected tissue. As the disease progresses rapidly and is associated with a high mortality, early diagnosis and timely implementation of antituberculous medication is critical.2,18 In our case, despite the delay in the etiologic diagnosis, the patient was treated properly with corticosteroids, cyclosporine and tuberculostatic agents. It is unknown whether TVR, a drug just recently started off-label in liver transplant recipients, may have contributed to the development of the HS. Unfortunately, as in many reported cases of HS, the outcome was unfavourable resulting in the death of the patient. Looking in retrospect to the course of this patient, we think that perhaps we should have stopped therapy at an earlier time point than done, once empirical therapy with antibiotics or increase in immunosuppression, particularly corticosteroids, did not result in clinical improvement. The choice of levofloxacin in the initial empirical antibiotic therapy was done because tuberculosis was part of the differential diagnosis. Stopping azathioprine or reducing prednisone doses at the start of antiviral therapy or during treatment was not (and should not) be considered given the “clear autoimmune component of liver injury” present in this patient.19,20
Conflict of InterestThe authors declare that they do not have anything to disclose regarding funding or conflict of interest with respect to this manuscript.



