




The MHC II deficiency is a rare autosomal recessive primary immunodeficiency syndrome with increased susceptibility to respiratory and gastrointestinal infections, failure to thrive and early mortality. This syndrome is caused by mutations in transcription regulators of the MHC II gene and results in development of blind lymphocytes due to the lack of indicatory MHC II molecules. Despite homogeneity of clinical manifestations of patients with MHC II deficiency, the genetic defects underlying this disease are heterogeneous. Herein, we report an Iranian patient with MHC II deficiency harbouring a novel mutation in RFXANK and novel misleading clinical features. He had ataxic gait and dysarthria from 30 months of age. Epidemiology, clinical and immunological features, therapeutic options and prognosis of patients with MHC II are reviewed in this paper.
MHC II deficiency is a rare autosomal recessive primary immunodeficiency syndrome (OMIM number: 209920) caused by mutations in the genes encoding transcription factors of MHC II genes. Most of the cases of MHC II deficiency have been reported from North African countries with a high prevalence of consanguineous marriages.1
Although MHC II deficiency is less profound than severe combined immunodeficiencies (SCID), it is a lethal condition that is associated with early childhood demise. Clinical manifestations of MHC II deficiency are similar to other combined immunodeficiencies and are comprised of early onset severe respiratory and gastrointestinal tract infections, failure to thrive (FTT), and early death. Other clinical manifestations of MHC II deficiency include septicaemia, neurological symptoms, and rarely autoimmune cytopenia.2
CD4+ lymphopenia, reversed CD4+/CD8+ ratio, and decreased level of one or more immunoglobulin subclasses are suggestive of MHC II deficiency, particularly when human immunodeficiency virus infection is excluded. Impaired antigen-specific antibody production and delayed-type hypersensitivity are other immunological features of MHC II deficiency. Although the definitive diagnosis is made by genetic analysis, the diagnosis is usually made based on the absence or limited expression of MHC II molecules on immune cells using flow-cytometry.
MHC II deficiency is categorised into four groups of A to D, based on the affected genes; RFXANK (Regulatory factor X associated protein containing ankyrin repeat), RFX5 (Regulatory factor X-5), RFXAP (Regulatory factor associated protein), and CIITA (class II transactivator). The first three genes’ products bind to the X-box of the MHC II promoter thereby recruiting a general transcription factor CIITA and triggering the transcription of MHCII gene (Fig. 1). Apart from its specific transactivation, CIITA functionally replaces a basal transcription factor TAF1 (TATA-Box Binding Protein Associated Factor 1), recruits the PIC (preinitiation complex) to the core promoter and with its acetyltransferase activity assures a proper transcription initiation of MHC I and II genes.3

Here we report a case with a novel homozygous mutation in the RFXANK gene followed by a review of the literature about related mutations found in patients with MHC II deficiency.
ReportHerein, we report a five-year-old Iranian boy from a consanguineous family (Fig. 2) who was initially diagnosed with SCID and was under treatment for SCID. He was referred to this centre because of the normal immunoglobulin (Ig) levels which were inconsistent with the SCID diagnosis. He had experienced episodes of fever, protracted diarrhoea and recurrent aphthous lesions in his mouth from four months of age. He had not been vaccinated after four months of age due to the early diagnosis of SCID. He had later developed severe upper respiratory infections. He suffered from FTT and developmental delay as he did not reach the growth and developmental milestones on the expected timeline. He had sat since 18 months of age and had walked without support when he was 30 months. His gait was ataxic and he had dysarthria.

Providing the patient's familial history, the patient's mother gave birth to a dead female foetus. Unfortunately, there is no evidence of the foetus's genetic analysis. Moreover, the patient's uncle (mother's brother) died of a disease with similar symptoms in his childhood. The patient's pedigree is illustrated in Fig. 2.
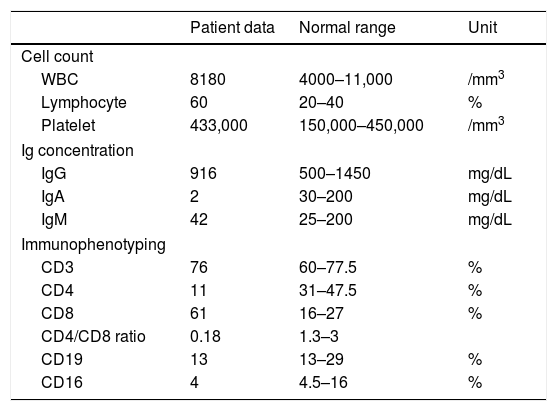
Laboratory findings revealed low CD4+ T cell count and inverted CD4/CD8 ratio (Table 1). Although total Ig levels were normal, decreased IgA level was detected. Increased antibody level observed after pneumococci vaccination confirmed normal immunity response to conjugate vaccines. Flow cytometry revealed no expression of MHC II antigen on monocytes and on B-cells. Following these results, the diagnosis of MHC II deficiency was made.
Immunological features of the patient with the associated normal range in this patient's age group. white blood cells (WBC), immunoglobulin (Ig).
| Patient data | Normal range | Unit | |
|---|---|---|---|
| Cell count | |||
| WBC | 8180 | 4000–11,000 | /mm3 |
| Lymphocyte | 60 | 20–40 | % |
| Platelet | 433,000 | 150,000–450,000 | /mm3 |
| Ig concentration | |||
| IgG | 916 | 500–1450 | mg/dL |
| IgA | 2 | 30–200 | mg/dL |
| IgM | 42 | 25–200 | mg/dL |
| Immunophenotyping | |||
| CD3 | 76 | 60–77.5 | % |
| CD4 | 11 | 31–47.5 | % |
| CD8 | 61 | 16–27 | % |
| CD4/CD8 ratio | 0.18 | 1.3–3 | |
| CD19 | 13 | 13–29 | % |
| CD16 | 4 | 4.5–16 | % |
Next generation sequencing targeting the known primary-immunodeficiency genes from International Union of Immunological Societies 20144 revealed a homozygous mutation in RFXANK gene on chromosome 19p12. The mutation is a G to A substitution in TGG codon for W188, leading to a premature stop codon in exon 3. Combined Annotation Dependent Depletion (CADD) score, which is a measure of the deleteriousness of single nucleotide variants, is 35.00 for this mutation. The mutation was confirmed by Sanger analysis and it showed a perfect segregation with the parents.
Even though the patient was admitted and intravenous antibiotic treatment was initiated, he died of pneumonia and septicaemia.
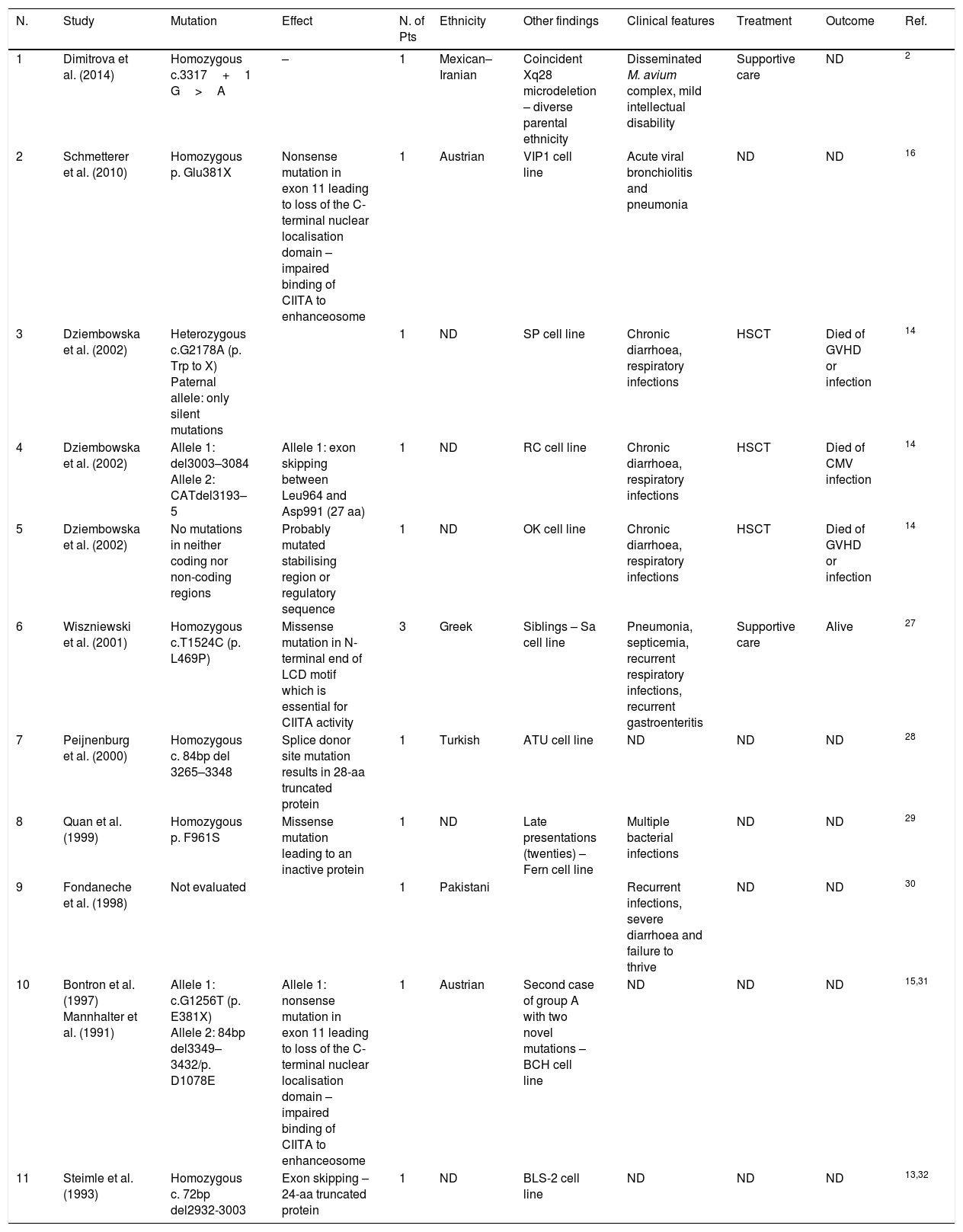
DiscussionMHC II deficiency is classified according to the MHC II molecule expression after pairwise fusion of different patient cell lines. The complementation groups were named by their order of discovery. There have been four complementation groups since 1997, when Durand et al. introduced MHC II deficiency group D with defects in RFXAP gene.5 More than half of the patients with MHC II deficiency have mutations in RFXANK, which are categorised as Group B. In this paper, we reported a novel mutation in RFXANK gene that caused MHC II deficiency in an Iranian boy. Moreover, we performed a systematic literature review to find all reported mutations causing MHC II deficiency. The authors searched English language literature using PubMed search engine with key words of “MHC class II deficiency” and “Bare lymphocyte syndrome” up to August 2016. The data were extracted from those studies with a reported mutation in patients or cell lines. Cases without mutation analysis were excluded from this study. Here, we present each study with associated mutations, ethnicity and clinical symptoms in four tables regarding each group individually. The results of this systematic search have been presented in Tables 2–5.
Group A mutations. Not determined (ND), number (N.), patients (Pts).
| N. | Study | Mutation | Effect | N. of Pts | Ethnicity | Other findings | Clinical features | Treatment | Outcome | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Dimitrova et al. (2014) | Homozygous c.3317+1 G>A | – | 1 | Mexican–Iranian | Coincident Xq28 microdeletion – diverse parental ethnicity | Disseminated M. avium complex, mild intellectual disability | Supportive care | ND | 2 |
| 2 | Schmetterer et al. (2010) | Homozygous p. Glu381X | Nonsense mutation in exon 11 leading to loss of the C-terminal nuclear localisation domain – impaired binding of CIITA to enhanceosome | 1 | Austrian | VIP1 cell line | Acute viral bronchiolitis and pneumonia | ND | ND | 16 |
| 3 | Dziembowska et al. (2002) | Heterozygous c.G2178A (p. Trp to X) Paternal allele: only silent mutations | 1 | ND | SP cell line | Chronic diarrhoea, respiratory infections | HSCT | Died of GVHD or infection | 14 | |
| 4 | Dziembowska et al. (2002) | Allele 1: del3003–3084 Allele 2: CATdel3193–5 | Allele 1: exon skipping between Leu964 and Asp991 (27 aa) | 1 | ND | RC cell line | Chronic diarrhoea, respiratory infections | HSCT | Died of CMV infection | 14 |
| 5 | Dziembowska et al. (2002) | No mutations in neither coding nor non-coding regions | Probably mutated stabilising region or regulatory sequence | 1 | ND | OK cell line | Chronic diarrhoea, respiratory infections | HSCT | Died of GVHD or infection | 14 |
| 6 | Wiszniewski et al. (2001) | Homozygous c.T1524C (p. L469P) | Missense mutation in N-terminal end of LCD motif which is essential for CIITA activity | 3 | Greek | Siblings – Sa cell line | Pneumonia, septicemia, recurrent respiratory infections, recurrent gastroenteritis | Supportive care | Alive | 27 |
| 7 | Peijnenburg et al. (2000) | Homozygous c. 84bp del 3265–3348 | Splice donor site mutation results in 28-aa truncated protein | 1 | Turkish | ATU cell line | ND | ND | ND | 28 |
| 8 | Quan et al. (1999) | Homozygous p. F961S | Missense mutation leading to an inactive protein | 1 | ND | Late presentations (twenties) – Fern cell line | Multiple bacterial infections | ND | ND | 29 |
| 9 | Fondaneche et al. (1998) | Not evaluated | 1 | Pakistani | Recurrent infections, severe diarrhoea and failure to thrive | ND | ND | 30 | ||
| 10 | Bontron et al. (1997) Mannhalter et al. (1991) | Allele 1: c.G1256T (p. E381X) Allele 2: 84bp del3349–3432/p. D1078E | Allele 1: nonsense mutation in exon 11 leading to loss of the C-terminal nuclear localisation domain – impaired binding of CIITA to enhanceosome | 1 | Austrian | Second case of group A with two novel mutations – BCH cell line | ND | ND | ND | 15,31 |
| 11 | Steimle et al. (1993) | Homozygous c. 72bp del2932-3003 | Exon skipping – 24-aa truncated protein | 1 | ND | BLS-2 cell line | ND | ND | ND | 13,32 |
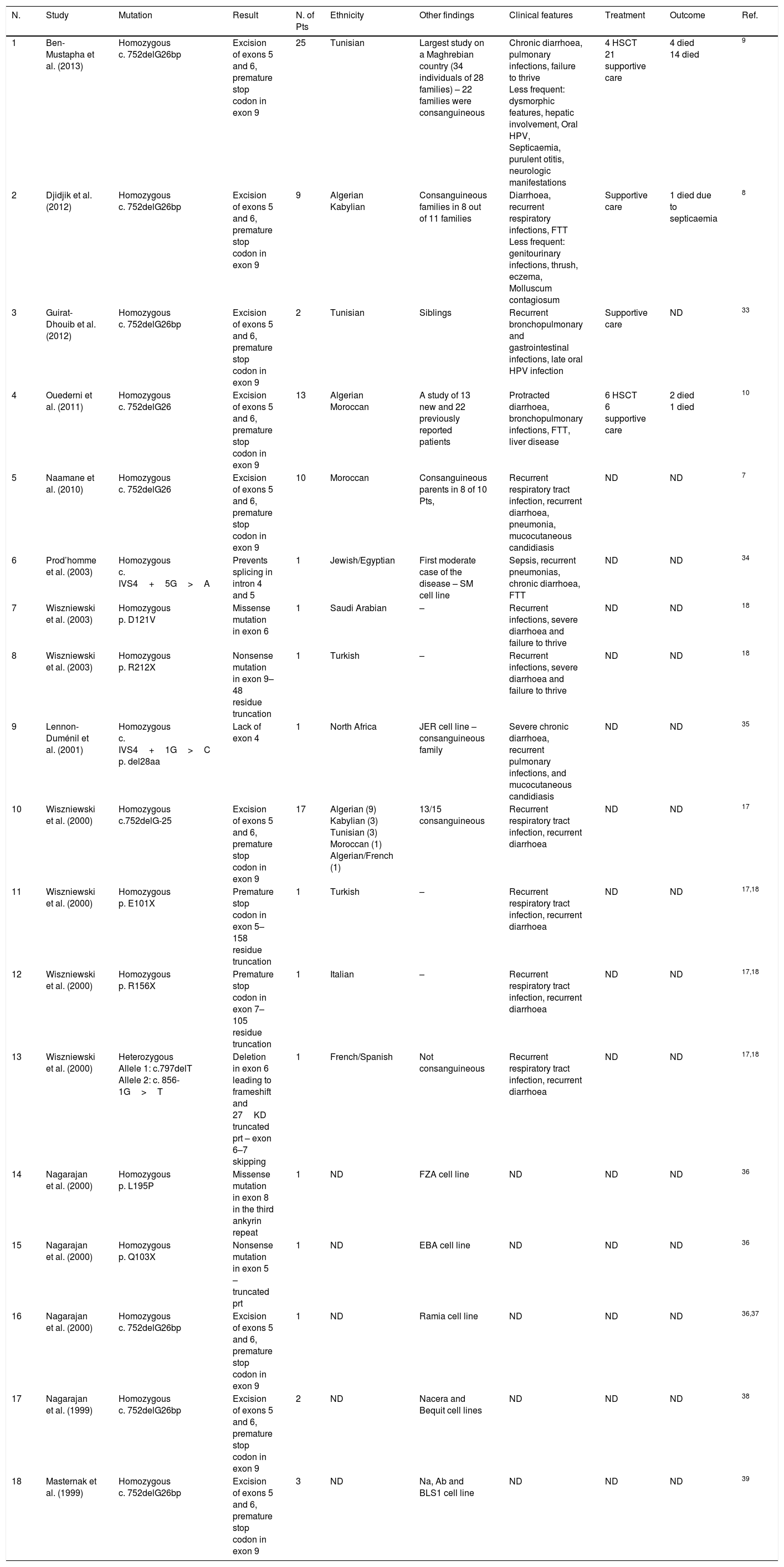
Group B mutations. Not determined (ND), failure to thrive (FTT), HPV (human papilloma virus), number (N.), patients (Pts).
| N. | Study | Mutation | Result | N. of Pts | Ethnicity | Other findings | Clinical features | Treatment | Outcome | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Ben-Mustapha et al. (2013) | Homozygous c. 752delG26bp | Excision of exons 5 and 6, premature stop codon in exon 9 | 25 | Tunisian | Largest study on a Maghrebian country (34 individuals of 28 families) – 22 families were consanguineous | Chronic diarrhoea, pulmonary infections, failure to thrive Less frequent: dysmorphic features, hepatic involvement, Oral HPV, Septicaemia, purulent otitis, neurologic manifestations | 4 HSCT 21 supportive care | 4 died 14 died | 9 |
| 2 | Djidjik et al. (2012) | Homozygous c. 752delG26bp | Excision of exons 5 and 6, premature stop codon in exon 9 | 9 | Algerian Kabylian | Consanguineous families in 8 out of 11 families | Diarrhoea, recurrent respiratory infections, FTT Less frequent: genitourinary infections, thrush, eczema, Molluscum contagiosum | Supportive care | 1 died due to septicaemia | 8 |
| 3 | Guirat-Dhouib et al. (2012) | Homozygous c. 752delG26bp | Excision of exons 5 and 6, premature stop codon in exon 9 | 2 | Tunisian | Siblings | Recurrent bronchopulmonary and gastrointestinal infections, late oral HPV infection | Supportive care | ND | 33 |
| 4 | Ouederni et al. (2011) | Homozygous c. 752delG26 | Excision of exons 5 and 6, premature stop codon in exon 9 | 13 | Algerian Moroccan | A study of 13 new and 22 previously reported patients | Protracted diarrhoea, bronchopulmonary infections, FTT, liver disease | 6 HSCT 6 supportive care | 2 died 1 died | 10 |
| 5 | Naamane et al. (2010) | Homozygous c. 752delG26 | Excision of exons 5 and 6, premature stop codon in exon 9 | 10 | Moroccan | Consanguineous parents in 8 of 10 Pts, | Recurrent respiratory tract infection, recurrent diarrhoea, pneumonia, mucocutaneous candidiasis | ND | ND | 7 |
| 6 | Prod’homme et al. (2003) | Homozygous c. IVS4+5G>A | Prevents splicing in intron 4 and 5 | 1 | Jewish/Egyptian | First moderate case of the disease – SM cell line | Sepsis, recurrent pneumonias, chronic diarrhoea, FTT | ND | ND | 34 |
| 7 | Wiszniewski et al. (2003) | Homozygous p. D121V | Missense mutation in exon 6 | 1 | Saudi Arabian | – | Recurrent infections, severe diarrhoea and failure to thrive | ND | ND | 18 |
| 8 | Wiszniewski et al. (2003) | Homozygous p. R212X | Nonsense mutation in exon 9–48 residue truncation | 1 | Turkish | – | Recurrent infections, severe diarrhoea and failure to thrive | ND | ND | 18 |
| 9 | Lennon-Duménil et al. (2001) | Homozygous c. IVS4+1G>C p. del28aa | Lack of exon 4 | 1 | North Africa | JER cell line – consanguineous family | Severe chronic diarrhoea, recurrent pulmonary infections, and mucocutaneous candidiasis | ND | ND | 35 |
| 10 | Wiszniewski et al. (2000) | Homozygous c.752delG-25 | Excision of exons 5 and 6, premature stop codon in exon 9 | 17 | Algerian (9) Kabylian (3) Tunisian (3) Moroccan (1) Algerian/French (1) | 13/15 consanguineous | Recurrent respiratory tract infection, recurrent diarrhoea | ND | ND | 17 |
| 11 | Wiszniewski et al. (2000) | Homozygous p. E101X | Premature stop codon in exon 5–158 residue truncation | 1 | Turkish | – | Recurrent respiratory tract infection, recurrent diarrhoea | ND | ND | 17,18 |
| 12 | Wiszniewski et al. (2000) | Homozygous p. R156X | Premature stop codon in exon 7–105 residue truncation | 1 | Italian | – | Recurrent respiratory tract infection, recurrent diarrhoea | ND | ND | 17,18 |
| 13 | Wiszniewski et al. (2000) | Heterozygous Allele 1: c.797delT Allele 2: c. 856-1G>T | Deletion in exon 6 leading to frameshift and 27KD truncated prt – exon 6–7 skipping | 1 | French/Spanish | Not consanguineous | Recurrent respiratory tract infection, recurrent diarrhoea | ND | ND | 17,18 |
| 14 | Nagarajan et al. (2000) | Homozygous p. L195P | Missense mutation in exon 8 in the third ankyrin repeat | 1 | ND | FZA cell line | ND | ND | ND | 36 |
| 15 | Nagarajan et al. (2000) | Homozygous p. Q103X | Nonsense mutation in exon 5 – truncated prt | 1 | ND | EBA cell line | ND | ND | ND | 36 |
| 16 | Nagarajan et al. (2000) | Homozygous c. 752delG26bp | Excision of exons 5 and 6, premature stop codon in exon 9 | 1 | ND | Ramia cell line | ND | ND | ND | 36,37 |
| 17 | Nagarajan et al. (1999) | Homozygous c. 752delG26bp | Excision of exons 5 and 6, premature stop codon in exon 9 | 2 | ND | Nacera and Bequit cell lines | ND | ND | ND | 38 |
| 18 | Masternak et al. (1999) | Homozygous c. 752delG26bp | Excision of exons 5 and 6, premature stop codon in exon 9 | 3 | ND | Na, Ab and BLS1 cell line | ND | ND | ND | 39 |
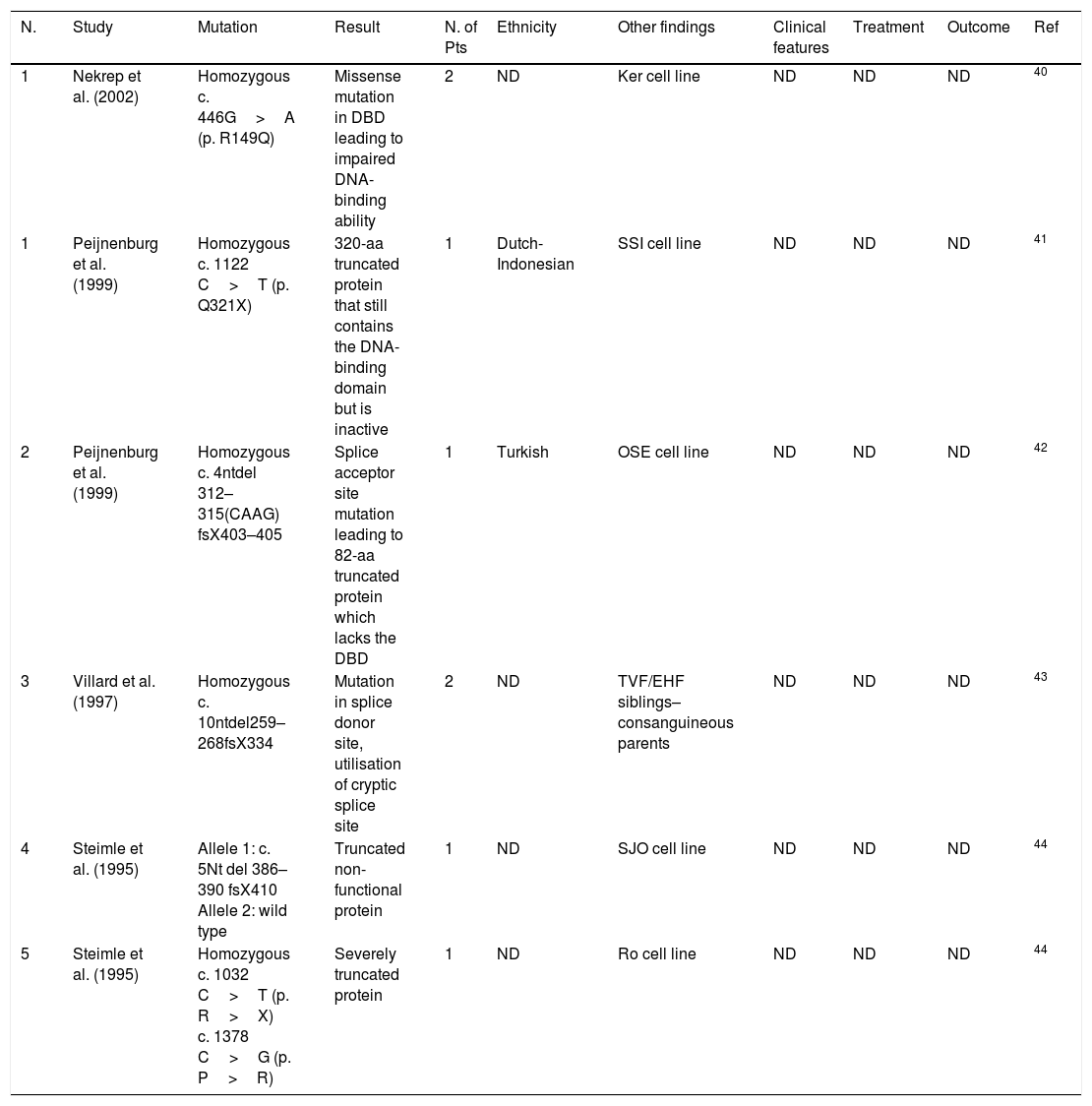
Group C mutations. DNA binding domain (DBD), not determined (ND), number (N.), patients (Pts).
| N. | Study | Mutation | Result | N. of Pts | Ethnicity | Other findings | Clinical features | Treatment | Outcome | Ref |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Nekrep et al. (2002) | Homozygous c. 446G>A (p. R149Q) | Missense mutation in DBD leading to impaired DNA-binding ability | 2 | ND | Ker cell line | ND | ND | ND | 40 |
| 1 | Peijnenburg et al. (1999) | Homozygous c. 1122 C>T (p. Q321X) | 320-aa truncated protein that still contains the DNA-binding domain but is inactive | 1 | Dutch-Indonesian | SSI cell line | ND | ND | ND | 41 |
| 2 | Peijnenburg et al. (1999) | Homozygous c. 4ntdel 312–315(CAAG) fsX403–405 | Splice acceptor site mutation leading to 82-aa truncated protein which lacks the DBD | 1 | Turkish | OSE cell line | ND | ND | ND | 42 |
| 3 | Villard et al. (1997) | Homozygous c. 10ntdel259–268fsX334 | Mutation in splice donor site, utilisation of cryptic splice site | 2 | ND | TVF/EHF siblings–consanguineous parents | ND | ND | ND | 43 |
| 4 | Steimle et al. (1995) | Allele 1: c. 5Nt del 386–390 fsX410 Allele 2: wild type | Truncated non-functional protein | 1 | ND | SJO cell line | ND | ND | ND | 44 |
| 5 | Steimle et al. (1995) | Homozygous c. 1032 C>T (p. R>X) c. 1378 C>G (p. P>R) | Severely truncated protein | 1 | ND | Ro cell line | ND | ND | ND | 44 |
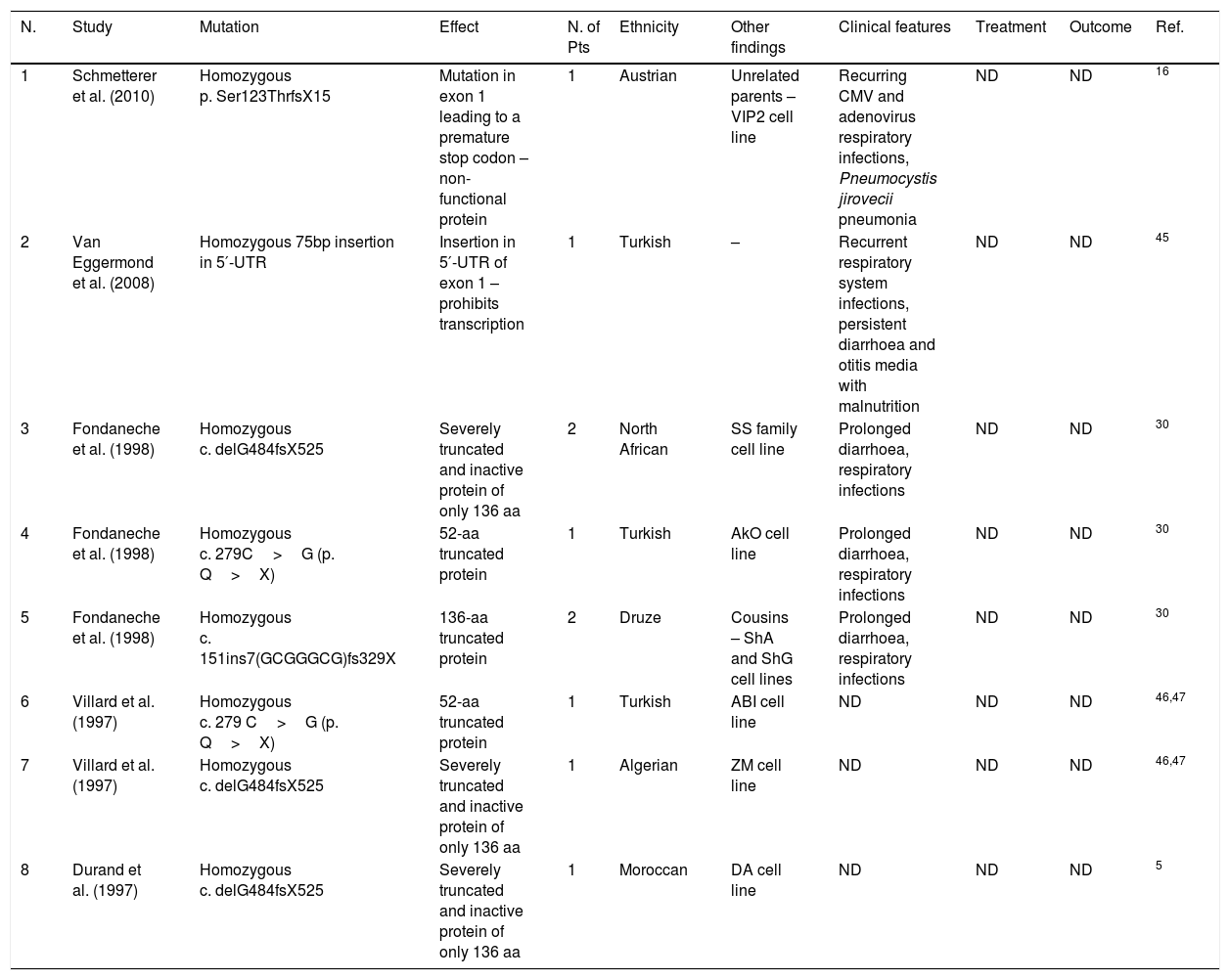
Group D mutations. Amino acid (aa), not determined (ND), cytomegalovirus (CMV), number (N.), patients (Pts).
| N. | Study | Mutation | Effect | N. of Pts | Ethnicity | Other findings | Clinical features | Treatment | Outcome | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Schmetterer et al. (2010) | Homozygous p. Ser123ThrfsX15 | Mutation in exon 1 leading to a premature stop codon – non-functional protein | 1 | Austrian | Unrelated parents – VIP2 cell line | Recurring CMV and adenovirus respiratory infections, Pneumocystis jirovecii pneumonia | ND | ND | 16 |
| 2 | Van Eggermond et al. (2008) | Homozygous 75bp insertion in 5′-UTR | Insertion in 5′-UTR of exon 1 – prohibits transcription | 1 | Turkish | – | Recurrent respiratory system infections, persistent diarrhoea and otitis media with malnutrition | ND | ND | 45 |
| 3 | Fondaneche et al. (1998) | Homozygous c. delG484fsX525 | Severely truncated and inactive protein of only 136 aa | 2 | North African | SS family cell line | Prolonged diarrhoea, respiratory infections | ND | ND | 30 |
| 4 | Fondaneche et al. (1998) | Homozygous c. 279C>G (p. Q>X) | 52-aa truncated protein | 1 | Turkish | AkO cell line | Prolonged diarrhoea, respiratory infections | ND | ND | 30 |
| 5 | Fondaneche et al. (1998) | Homozygous c. 151ins7(GCGGGCG)fs329X | 136-aa truncated protein | 2 | Druze | Cousins – ShA and ShG cell lines | Prolonged diarrhoea, respiratory infections | ND | ND | 30 |
| 6 | Villard et al. (1997) | Homozygous c. 279 C>G (p. Q>X) | 52-aa truncated protein | 1 | Turkish | ABI cell line | ND | ND | ND | 46,47 |
| 7 | Villard et al. (1997) | Homozygous c. delG484fsX525 | Severely truncated and inactive protein of only 136 aa | 1 | Algerian | ZM cell line | ND | ND | ND | 46,47 |
| 8 | Durand et al. (1997) | Homozygous c. delG484fsX525 | Severely truncated and inactive protein of only 136 aa | 1 | Moroccan | DA cell line | ND | ND | ND | 5 |
The geographic distribution of MHC II deficiency is confined to areas with a high prevalence of consanguineous marriages. Thus MHC II deficiency is more prevalent in middle European countries, Middle Eastern countries and African countries, with the highest prevalence observed in Mediterranean areas.6 Of 103 reported patients with MHCII whose ethnicity is known, 94 (91.2%) come from Asian, Middle-East or African families (Tables 2–5). These regions have a high prevalence of consanguineous marriages, which increases the incidence of autosomal recessive disorders due to the limited gene pool. Group B MHC II deficiency is more concentrated in North African areas and Group A seems to be more prevalent in European countries. Herein we report the first Iranian patient with group B MHC II deficiency.
Although MHC II deficiency is considered as a related SCID disorder, almost none of the patients developed the first manifestations around birth. There is a considerable time gap between the onset of infections and the diagnosis. This diagnostic gap has been elucidated in several studies on patients with group B MHC II deficiency. The mean age at the time of the first presentation was 4.2–10 months of age7–10 and the mean age at the time of diagnosis was 21–42 months of age.8–10 Thus, there has been a 17–32-month diagnostic gap in reported cases. This diagnostic gap may be due to the non-specific or less severe manifestations at the beginning of the disease.
In this patient, despite the early onset of disease at four months of age, the syndrome was diagnosed at five years of age, which is remarkably higher than the mean age of diagnosis in other studies and that may be because of the patient's unusual clinical presentation, which is discussed later in the text.
Clinical findingsSimilarly, in all four complementation groups we did not notice any significant difference in clinical manifestations. Almost all of the patients develop respiratory and gastrointestinal infections several months prior to making the diagnosis of MHC II deficiency. Diffuse and severe infections of gastrointestinal, cutaneous and respiratory origin with rare pathogens are common findings in patients with MHC II deficiency. Other less frequent manifestations are FTT, intellectual disability, transaminitis and neurological symptoms, septicaemia and autoimmune cytopenias. The non-specific clinical presentation of the disease may be a reason for the prolonged lag of diagnosis from the onset of manifestations.
The clinical features in group B patients are more diverse. There are some unique presentations which are not reported in other groups e.g. dysmorphic features, transaminitis, oral HPV and candidiasis, thrush, purulent otitis, neurological manifestations, genitourinary infections, eczema and Molluscum contagiosum.8,9 The exclusive manifestation of these symptoms in patients with group B MHC II deficiency should be further studied.
In addition to the classical clinical features observed in patients with MHC II deficiency, the patient reported here had several new manifestations that may be worth studying more carefully for their relevance to the BLS syndrome. One major presentation was recurrent oral aphthous, which have not yet been reported in the literature. Previously, oral ulcers were reported in a five-year-old boy with MHC II deficiency from a non-consanguineous marriage between an Iranian father and an American-Mexican mother.2 Moreover, the patient suffered from two neurological disabilities, dysarthria and ataxic gait. These heterogeneous clinical features might be misleading in the early diagnosis of the disease.
Immunological featuresThere are no specific differences in laboratorial findings in patients of different groups. Although the common immunophenotype is CD4+ lymphopenia with reversed CD4/CD8 ratio,1 a patient with MHC II deficiency was reported to have normal CD4/CD8 ratio due to the decreased CD8+ count.11 The CD4+ lymphopenia is a result of impaired MHC II-dependant thymic maturation of T cells.1 Serum immunoglobulin subclasses are variable and may be normal, decreased or increased. The count of B cells and NK cells are normal in most cases. Since the immune response to foreign antigens is impaired, patients with MHC II deficiency fail to develop a proper active immunity by vaccination. However, the immunodeficiency is not severe enough to develop disseminated infections after vaccination. Thus, patients with MHC II deficiency are vaccinated according to the national vaccination programme in their country of residence. There is one reported case of disseminated BCGitis in a patient who was later diagnosed to have probable MHC II deficiency.12 Moreover, another patient of group A MHC II deficiency developed symptomatic varicella after vaccine administration.2
The main immunological findings in the presented patient were low CD4+ T cell count and inverted CD4/CD8 ratio. There was only a decline in IgA level and other immunoglobulin levels were in normal limits. Because of the unique presentation of the disease and early diagnosis of SCID, the patient was not vaccinated after four months of age. But during his referral to our centre, the patient showed normal response to conjugated vaccines due to the fourfold increase in anti-pneumococcal antibody after vaccination.
Mutation analysisThe list of mutations underlying MHC II deficiency is growing. It sheds light on the pathophysiology of disease and opens new windows to immunologists studying primary immunodeficiencies. Here we added a novel mutation in RFXANK gene, which is categorised as group B of MHC II deficiency. This mutation similar to others leads to decreased MHC II transcription and thereby MHC II expression by immune cells which are illustrated in Fig. 1. Identifications of genetic defects underlying MHC II deficiency can be of value for targeted gene therapy - which is a dream at the moment.
The first described MHC II deficiency disease-causing gene was CIITA,13 a 28-exon gene located on 16p13 locus. There are three isoforms of CIITA, all having a Leucine-rich motif (LRM) participating in protein–protein interactions. The LRM is located in residues 790–1115, 789–1114 and 299–530 of the three isoforms respectively. To the best of our knowledge, approximately 12 different mutations have been reported (Table 2). The mutations either lead to a premature stop codon resulting in a truncated protein or are missense mutations resulting in inactive proteins. Most of the defected CIITA proteins are unable to bind to the enhanceosome complex in the promoter of MHC II gene because the LRM is involved. There is only one patient with no mutations in the coding and non-coding areas of CIITA gene.14 The mutation is suspected to be in the regulatory areas upstream of the CIITA. There were two unrelated Austrian patients with the same nonsense p. E381X mutations, suggesting a possible association of this mutation with Austrian ethnicity.15,16 There was also one patient with a heterozygous mutation with only null mutations in one allele.14
Group B MHC II deficiency with mutations in RFXANK gene was the second to be discovered. RFXANK gene is located on 19p12 locus with 12 exons in its transcript. There are several isoforms of RFXANK proteins differing in the number of ankyrin repeats, which are the protein–protein interaction sites crucial for binding to other RFX proteins. Each repeat contains two antiparallel helices and a beta-hairpin and is stacked in a superhelical arrangement coupled with other repeats. Generally, there have been 11 reported mutations in group B BLS patients that affect exons 4–9 of the RFXANK. Except two missense mutations in exons 6 and 8, the remainder result in truncated proteins or excision of one or more exons. All of the reported mutations, either nonsense or splice-site mutations, result in proteins lacking either a part or all of the ankyrin repeats.
The most frequent mutation in group B MHC II deficiency patients is a 752delG26 leading to excision of exons 5 and 6 and a premature stop codon in exon 9. The founder effect of this mutation in North African patients with group B MHC II deficiency, had been first documented by Wiszniewski et al.17 The largest study that confirmed the founder effect of c. 752delG26 mutation in 34 Tunisian patients from 28 families was done by Ben-Mustapha et al.9 Another large study was performed on 20 MHC II deficiency patients, out of whom 17 patients harboured the common mutation 752delG26 and were originally from North African countries.17 The three remaining patients of the latter study, in addition to two more cases were studied elsewhere revealing six mutations consisting of four homozygous mutations and one patient with heterozygous mutation.18
The patient described here is the first patient with p. W188X mutation due to a G to A substitution, leading to a non-functional RFXANK gene product. This mutation is the first mutation affecting exon 3 and preceding the fourth ankyrin repeat, resulting in the truncated protein lacking ankyrin repeat 4 and 5. RFX5, the defected gene in group C patients, is located in locus 1q21 with a 12-exon transcript. There is a highly conserved DBD (DNA-binding domain) in RFX5 protein, located in 90–166 residues and 407–614 residues, which bind the ssDNA of the X-boxes prior to transcription. Out of the six mutations reported to now, five mutations result in a premature stop codon prior to the second DBD sequence and the other one is a missense mutation substituting an arginine in a DNA-binding surface sequence which leads to impaired binding of RFX5 to X-boxes.
RFXAP, a 3-exon gene located in 13q14 locus, was the last defected gene to be identified in a subgroup of MHC II deficiency patients. Durand et al. revealed the association of this gene with MHC II deficiency from an in vitro generated mutant, 6.1.6,5 which was mutagenised for studies on all antigens expressed by D locus of MHC II gene.19 There is a conserved sequence at the C-terminal end of the associated protein (140–243 residues) which is responsible for binding to DNA. Five different mutations have been discovered to now. Interestingly, two unrelated Turkish patients harboured the same mutation and four unrelated North African harboured the same c. delG484fsX525 mutation, proposing the prevalence of each mutation in the associated region. Four out of five reported mutations resulted in premature stop codon prior to the C-terminal binding domain of RFXAP protein. The other reported mutation in a Turkish patient is a 75bp insertion in 5′-UTR, leading to the inhibition of transcription.
TreatmentEven though the MHC II deficiency is not as severe as SCID, most patients fail to reach their puberty and die of severe infections, even on antibiotic treatment. Supportive care of these patients may help patients survive only up to early childhood.10 Patients with MHC II deficiency who do not undergo haematopoietic stem cell transplantation (HSCT) succumb to a complicated course and demise by a median age of four to five years.20 Although HSCT is considered as the only curative treatment, studies show a high rate of post-HSCT death because of severe infections, graft versus host diseases (GVHD) and organ failures.10,11,21,22 The main cause of the poor outcome of HSCT is GVHD. Even though the lymphocytes of the patients with MHC II deficiency do not express MHC II which is required for the rejection process, the residual immunity is still enough for developing rejection. The antigen presenting cells of the donor may have a role in activating recipient's T cells and in this regard, MHC II deficiency patients must have a HLA-matched donor.
Although studies found that myeloablative or reduced intensity conditioning can be used to achieve a better post-transplant prognosis,23 Siepermann et al. demonstrated a better prognosis in patients with non-myeloablative preconditioning. This is assumed to be associated with preservation of the immune function against existing infections prior to the transplant. Renella et al., reported a 50% rate of GVHDs higher than grade I in 14 patients with MHC II deficiency that had undergone HSCT.24 A study on 23 patients with MHC II deficiency reported a rate of about 50% (12/23) deaths one month to six years after HSCT, most of which happened immediately after the transplant due to severe GVHD or severe infections.10 In comparison, seven out of 12 patients on supportive care who did not undergo HSCT were alive at the end of this study. Siepermann et al. studied 68 patients that had been transplanted, 41 of whom were preconditioned with myeloablative regimen.20 Of them, 10 patients developed graft rejection and 42 patients died of GVHD or severe infections. Another study on 16 patients with MHC II deficiency who underwent HSCT demonstrated 31% (5/16) failure and 37% (6/16) GVHD, during six months after HSCT.25 More recently Ben-mustapha et al. reported 75% mortality in eight patients who had undergone HSCT mostly because of GVHD, with similar high rates of mortality of 70% in 26 patients who had not been transplanted mostly because of severe infections.9 Current literature on patients with definitive diagnosis of group B MHC II deficiency based on genetic analysis shows 43% (16/37) mortality following supportive care, and 69% (9/13) mortality following HSCT (Odds ratio; 2.9, 95%CI (0.76–11.3), p=0.120). Table 3 demonstrates detailed results on the outcome of HSCT and supportive care for patients with group B MHC II deficiency. To date, HSCT confers no additional survival benefit over supportive care for patients with group B MHC II deficiency.
Although the source of stem cells required for transplantation was bone marrow in most cases, a few studies reported umbilical cord blood transplantation (CBT). The extensive potency of cord blood cells to differentiate and populate bone marrow with a lower risk of contamination makes CBT a candidate therapeutic modality for patients with MHC II deficiency.20 However, the drawbacks of CBT are enormous, including, the high rate of graft failure and the limitation of therapeutic options for the donor in whom CBT has been failed.20 Acute GVHD,25 poor engraftment10 and remission20 are frequently observed following CBT for patients with MHC II deficiency. Thus, if stem cell transplantation is planned to be done for patients with MHC II deficiency, HSCT is still superior on CBT.
Patients should be observed carefully after HSCT, because CD4 lymphopenia may remain even after transplant due to lack of MHC II expression on thymic cells. Before transplantation, patients should be on prophylactic antibiotics and nutritional support and receive intravenous immunoglobulins (IVIG) in case of low serum immunoglobulins. IVIG and G-CSF has been used to increase serum immunoglobulin levels and lymphocyte counts but are not curative. Obviously, patients with MHC II deficiency need improvement of therapeutic modalities. Early diagnosis and treatment may improve the outcome of patients with MHC II deficiency by doing research in upcoming years. It is to be hoped that by better understanding the genetic background of patients with MHC II deficiency, a more efficient therapeutic strategy will be developed which targets the affected MHC II transcriptional regulatory factor genes.
CommentMHC II deficiency is a severe combined immunodeficiency syndrome with a protracted course. Typical manifestations include respiratory and gastrointestinal tract infections, diarrhoea, FTT, developmental delay and demise in early childhood. Diagnosis is usually made by complete blood cell counts (CBC), immunoglobulin assessment and flow cytometry which reveal decreased CD4+ counts and the absence of MHC II expression by immune cells. Despite the classical clinical presentation and developed diagnostic tools, the diagnostic gap is still an important factor underlying a poor outcome in these children. The prevalence of MHC II deficiency is highest in North Africa and Middle-East where low resources do not allow for newborn screening for such a primary immunodeficiencies. In western countries such as the United States, screening for SCID has been added to the national newborn screening programme but it also lacks identification of MHC II deficiency as a SCID related disorder.26 This issue may be addressed by modified newborn screening programmes for families with a consanguineous marriage or history of primary immunodeficiency in their first-degree relatives. Another problem here is that an appropriate screening tool has not yet been developed for MHC II deficiency. T-cell receptor excision circles (TRECs) which are by-products of T-cell receptor rearrangement are used to screen newborns for SCID. Patients with MHC II deficiency have T+ combined immunodeficiency and current TREC cut points used for SCID screening cannot identify MHC II deficiency as well.26 The question is now whether newborn screening for MHC II deficiency is mandatory and cost-effective in North-Africa and Middle-East or not and if so, how should it be done.
ConclusionHerein, we described a case of group B MHC II deficiency with a novel nonsense mutation in the third exon of RFXANK, resulting in excision of ankyrin repeats which are responsible for protein–protein interaction. The latter may result in defected interaction of RFXANK with other Regulatory factor X family proteins which are crucial for triggering the MHC II transcription. The previously reported mutations in RFXANK involve exon 4–9 and result in lack of the ankyrin repeats. The patient reported here manifested with unusual symptoms; he had developed recurrent aphthous lesions, dysarthria and ataxia. The unusual manifestations of the patient misled the physicians in diagnosing the disease and the patient died of disseminated infections, under antibiotic therapy. The immunological findings were the same as other reported cases and consisted of CD4+ lymphopenia, reversed CD4/CD8 ratio and decreased IgA concentration. We have provided a review of the reported MHC II deficiency cases to help understanding of the spectrum of epidemiological, clinical, immunological and genetic characteristics of the MHC II deficiency disease and the therapeutic outcomes. The diagnostic gap and the misleading nature of the disease may be the main reasons for the poor outcome of MHC II deficiency patients.
Ethical disclosuresConfidentiality of dataThe authors declare that they have followed the protocols of their work centre on the publication of patient data and that all the patients included in the study have received sufficient information and have given their informed consent in writing to participate in that study.
Right to privacy and informed consentThe authors have obtained the informed consent of the patients and/or subjects mentioned in the article. The author for correspondence is in possession of this document.
Protection of human subjects and animals in researchThe authors declare that the procedures followed were in accordance with the regulations of the responsible Clinical Research Ethics Committee and in accordance with those of the World Medical Association and the Helsinki Declaration.
Conflict of interestNothing to disclose.
This study was supported by Tehran University of Medical Sciences, grant no. 32345.
