





A día de hoy no existe una terapia médica eficaz para la poliquistosis hepática (PQH), considerándose tratamientos paliativos la punción quística con escleroterapia, la fenestración o la hepatectomía parcial. El trasplante ortotópico de hígado (TOH) es el tratamiento de elección para la PQH terminal, estando indicado en pacientes con síntomas limitantes no susceptibles de recibir tratamiento médico. Con la aplicación del sistema Model for End-Stage Liver Disease (MELD) es difícil determinar la prioridad en la lista de espera.
MétodosAnálisis retrospectivo de los TOH por PQH realizados consecutivamente en nuestro centro. Los criterios de inclusión para TOH en pacientes con síntomas limitantes fueron la presencia de poliquistosis bilateral (Gigot tipo iii) y la hepatomegalia masiva con un hígado remanente insuficiente que imposibilitara una hepatectomía. Se realizó de donante cadáver con técnica piggy-back sin by-pass veno-venoso.
ResultadosEntre abril de 1992 y abril de 2010 se realizaron 6 TOH, uno de ellos combinando trasplante hepatorrenal. La media de transfusión fue 3,25 concentrados de hematíes y 1.200 cc de plasma fresco congelado. El tiempo quirúrgico medio fue 299 min y 498 min en el hepatorrenal. No hubo mortalidad perioperatoria. La media de hospitalización fue 6,5 días, permaneciendo sanos todos los pacientes tras una media de seguimiento de 71 meses.
ConclusiónEl TOH ofrece una excelente supervivencia global. Los resultados son mejores cuando el trasplante se realiza de una manera precoz, por lo que estos pacientes deberían recibir una puntuación adicional para poder emplear el MELD como una escala válida.
There is currently no effective medical therapy for polycystic liver (PCL). Cyst puncture and sclerotherapy, cyst fenestration, or partial hepatic resections have been used as palliative treatments. Orthotopic liver transplantation (OLT) has become the treatment of choice for terminal PCL, being indicated in patients with limiting symptoms not susceptible to any other medical treatment. It is also difficult to determine the priority on the waiting list using the Model for End-Stage Liver Disease (MELD).
MethodsA retrospective analysis of OLT for PCL was conducted in our centre. Inclusion criteria were patients with limiting symptoms, bilateral cysts liver, and insufficient remaining liver. In all cases a deceased donor liver transplantation with piggy-back technique without veno-venous bypass was performed.
ResultsSix patients underwent liver transplantation for PCL between April 1992 and April 2010, one of them a combined liver-kidney transplantation. The mean intraoperative packed red blood cell transfusion was 3.25 L and fresh frozen plasma was 1.200 cc. Mean operation time was 299min, and 498min in the liver-kidney transplantation. There was no peri-operative mortality. The mean hospital stay was 6.5 days. All patients are healthy after a mean follow-up of 71 months.
ConclusionOLT offers an excellent overall survival. Results are better when OLT is performed early; thus these patients should receive additional points to be able to use the MELD score as a valid prioritisation system for waiting lists.
La poliquistosis hepática (PQH) es una enfermedad que puede ser debida a 2 alteraciones hereditarias diferentes. Una de ellas es la poliquistosis renal autosómica dominante y es causada por mutaciones de los genes PKD1 y PKD2. Se trata de una enfermedad multisistémica de alta penetrancia y de expresión variable. La mayoría de los pacientes con esta enfermedad presentan también poliquistosis hepática, aunque esta no suele ser la principal responsable de los síntomas1. La otra alteración es la enfermedad poliquística hepática, que es causada mayoritariamente por la mutación en PRKCSH o en SEC632,3 y que presenta quistes hepáticos de forma aislada4. En el epitelio de los quistes se expresan receptores de estrógenos, por lo que los síntomas son más frecuentes en mujeres5. En el momento del diagnóstico es importante incluir en el diagnóstico diferencial el cistoadenoma y el cistoadenocarcinoma, mediante TAC o RMN6, aunque la PQH no está asociada a un mayor riesgo de malignidad.
La sintomatología suele comenzar en la tercera o cuarta décadas de la vida y es debida al crecimiento de los quistes, por lo que es rara su presentación en la infancia. Aunque en la mayoría de los casos la capacidad de síntesis hepática está conservada, los pacientes pueden presentar disnea, dolor abdominal o saciedad precoz, por el efecto masa causado. En los casos más evolucionados aparece malnutrición7–9. Los síntomas se ven exacerbados cuando existe compresión de la vena porta, de la cava inferior o de la vía biliar extrahepática. Las principales complicaciones de los quistes son la rotura, la infección o el sangrado, siendo más frecuente en los casos que necesitan hemodiálisis10,11. Los síntomas se presentan cuando ya hay establecida una poliquistosis masiva, en los que la ratio quiste/parénquima suele ser > 112.
La PQH no suele requerir tratamiento. Cuando comienza a dar síntomas, se precisa disminuir el volumen quístico y hepático, sin que exista una terapia médica eficaz para ello. En función del paciente y del tamaño, localización y número de quistes, se puede optar por distintas alternativas. La menos invasiva es la aspiración guiada por ecografía o TAC del contenido de los quistes y la posterior instilación de agentes esclerosantes para evitar su recidiva13. Cuando esto no es suficiente, se requieren fenestraciones quísticas laparoscópicas o abiertas, que tienen asociadas un mayor riesgo de complicaciones como sangrado, fuga biliar, ascitis o derrame pleural14–16. Otra opción quirúrgica es la resección hepática parcial, que presenta altas tasas de morbilidad y tiene la dificultad añadida de tener que diseñar una línea de resección sobre un hígado deformado por los quistes y con riesgo de conservar escaso parénquima hepático sano remanente17. Además, las hepatectomías crean adherencias que dificultarían un posible trasplante hepático posterior, por lo que esta cirugía se debería limitar a los pacientes en los que la fenestración no es posible o que no sean candidatos a trasplante18. En cualquiera de estos casos se trata de soluciones paliativas, no curativas.
En los últimos años, el trasplante ortotópico de hígado (TOH) se ha convertido en el tratamiento de elección de la enfermedad hepática terminal por PQH, siendo la única opción curativa, junto con el trasplante hepático de donante vivo19,20. En ocasiones es necesario realizarlo de manera conjunta con el trasplante renal9,21,22. Desde que se realizó el primer TOH por PQH en 198823, la morbimortalidad se ha reducido gracias a un mejor manejo de la inmunosupresión y de los cuidados perioperatorios. Está indicado en pacientes sintomáticos, con gran disminución de la calidad de vida y no susceptibles de otro tratamiento médico. La clasificación de Gigot facilita la planificación del tratamiento idóneo en función de las características de los quistes: tipo i: pocos y grandes; tipo ii: múltiples y de tamaño intermedio; tipo iii: múltiples de tamaño pequeño e intermedio24.
MétodosSe realiza un análisis retrospectivo de los TOH llevados a cabo en nuestro centro por PQH. Los criterios de inclusión para TOH en pacientes con síntomas limitantes fueron la presencia de poliquistosis bilateral (Gigot tipo iii) y la hepatomegalia masiva con un hígado remanente insuficiente que imposibilitara una hepatectomía. En todos los casos se realizó TOH de donante cadáver siguiendo la técnica de piggy-back. En ninguna paciente se realizó by-pass veno-venoso. En todos los casos se administró una pauta personalizada de inmunosupresión, basada en tacrolimus, micofenolato mofetil y corticoides.
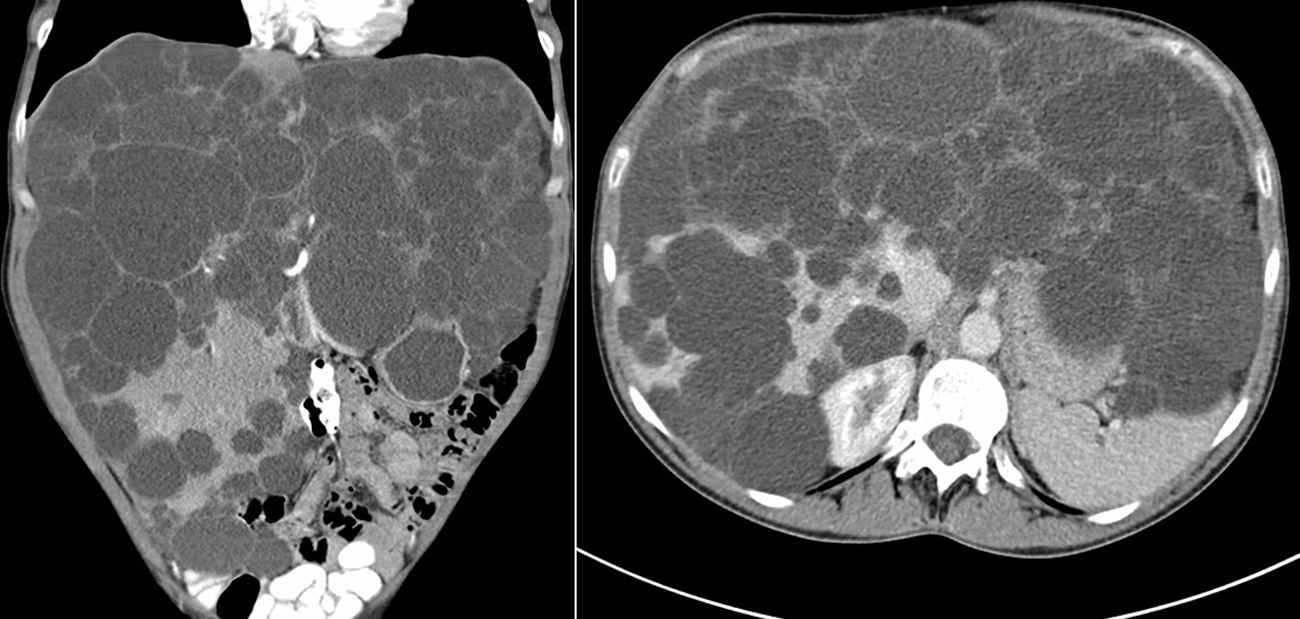
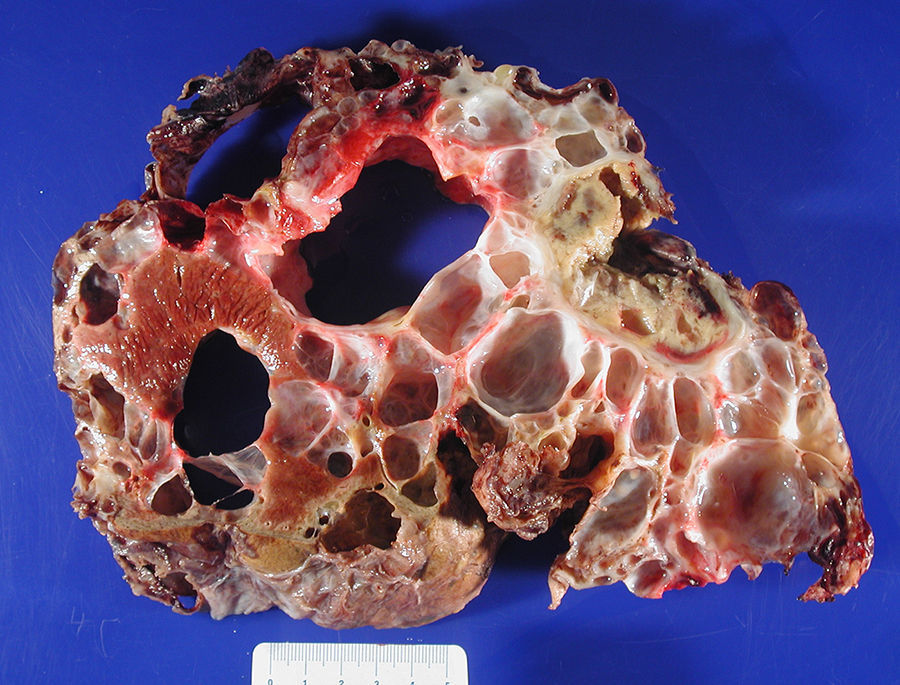
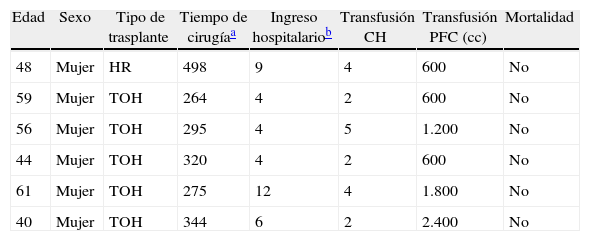
ResultadosEntre abril de 1992 y abril de 2010 se han realizado en nuestro centro 6 TOH por PQH. Los datos demográficos se recogen en la tabla 1. Todas las pacientes trasplantadas cumplían los criterios de inclusión en el momento de la realización del TOH y padecían síntomas progresivos durante, al menos, 5 años, principalmente en forma de plenitud posprandial, saciedad precoz, hepatomegalia, disnea y ascitis refractaria. En una paciente, que presentaba insuficiencia renal terminal en programa de hemodiálisis, se realizó doble trasplante hepatorrenal, sin asociar nefrectomía en el acto quirúrgico. La figura 1 muestra la imagen radiológica de una de las pacientes trasplantadas. La media de transfusión de concentrados de hematíes fue de 3,25 (rango: 2-5) y la de plasma fresco congelado fue de 1.200 cc (600-2.400). En uno de los casos también se transfundió una unidad de plaquetas. El tiempo operatorio fue de 299 min (264-344) en el TOH y de 498 en el doble hepatorrenal. El peso medio del hígado explantado fue de 4.112g (2.870-5.725), aunque en todos los casos se realizó fenestración quística intraoperatoria para facilitar la extracción. En la figura 2 se observa la imagen macroscópica de uno de los hígados resecados. No hubo mortalidad operatoria. La estancia hospitalaria fue de 6,5 (4-12) días, con un ingreso medio en la Unidad de Cuidados Intensivos de 1,5 (1-3) días. No fue reintervenido ningún paciente. Una paciente presentó estenosis de la anastomosis de la vena cava suprahepática, tratada con éxito mediante endoprótesis vascular, y presentó una pequeña eventración corregida quirúrgicamente. Otra presentó infección por CMV, con buena respuesta a tratamiento con ganciclovir. Todas las pacientes se encuentran sanas tras una media de seguimiento de 71 meses (32-246).
Características de los trasplantes
| Edad | Sexo | Tipo de trasplante | Tiempo de cirugíaa | Ingreso hospitalariob | Transfusión CH | Transfusión PFC (cc) | Mortalidad |
| 48 | Mujer | HR | 498 | 9 | 4 | 600 | No |
| 59 | Mujer | TOH | 264 | 4 | 2 | 600 | No |
| 56 | Mujer | TOH | 295 | 4 | 5 | 1.200 | No |
| 44 | Mujer | TOH | 320 | 4 | 2 | 600 | No |
| 61 | Mujer | TOH | 275 | 12 | 4 | 1.800 | No |
| 40 | Mujer | TOH | 344 | 6 | 2 | 2.400 | No |
CH: concentrados de hematíes; HR: hepatorrenal; PFC: plasma fresco congelado; TOH: trasplante ortotópico de hígado.


La PQH es una enfermedad rara con una prevalencia del 0,05-0,13% en series de autopsias25. En España la indicación de TOH debida a tumores benignos, como la PQH, es menor de 0,6%26. En el Registro Europeo de Trasplante Hepático, la PQH representa menos del 1%27. En nuestro centro se realizaron 354 TOH durante este periodo de tiempo, por lo que la PQH supone un 1,7%.
Aunque es considerada una entidad benigna, puede ser una fuente importante de morbilidad, e incluso de mortalidad en los casos más evolucionados. No existe un tratamiento médico apropiado, y las soluciones invasivas como la descompresión quística, la aspiración y esclerosis o la fenestración quirúrgica tan solo alivian los síntomas de una manera temporal en los casos de escasos quistes de gran volumen, pero no en los de múltiples quistes pequeños17,28,29. Existe controversia entre la elección de TOH o hepatectomía en este grupo de pacientes. La hepatectomía en la PQH está asociada con una mayor tasa de morbilidad debido a fuga biliar, ascitis postoperatoria, infecciones bacterianas y deterioro de la función renal. La hepatectomía debería ser el tratamiento de elección en pacientes sin alteraciones nutricionales y con una función renal conservada o levemente disminuida en los casos en que el hígado remanente sea mayor del 30% y teniendo en cuenta la distribución de los quistes y de la vascularización sectorial30,31.
El TOH se ha convertido en el tratamiento de elección en los casos de afectación hepática terminal y, en las últimas 2 décadas, ha alcanzado unos cada vez mejores porcentajes de supervivencia, tanto del injerto como del paciente, consiguiendo un gran alivio sintomático20,32–34. La principal indicación para el TOH es la presencia de síntomas limitantes con afectación quística masiva35, aunque, con la aplicación del sistema Model for End-Stage Liver Disease (MELD), es difícil determinar la prioridad en la lista de espera. En ese tiempo muchos pacientes experimentan un aumento de la distensión abdominal y del dolor, aunque la malnutrición suele ser posterior. Es controvertido el TOH en la PQH puesto que no existe ningún síntoma ni signo determinante para posicionar al paciente en la lista de espera36. A esto se suma la especial dificultad técnica de la intervención en la PQH, debida al gran tamaño hepático, a los procedimientos invasivos realizados previamente y al desplazamiento de estructuras vasculares y de órganos vecinos. Esta dificultad se asocia a una mayor pérdida sanguínea, con la consecuente morbilidad añadida37.
Todas nuestras intervenciones se llevaron a cabo con preservación de la vena cava inferior del receptor (piggy-back) y sin by-pass veno-venoso. Durante la operación fue necesaria la aspiración y fenestración quística para mejorar la visualización de las estructuras vasculares y permitir la movilización hepática, sin haber incidencia de perforación de órganos adyacentes. La hepatectomía se convierte en el principal reto quirúrgico y es donde existe controversia sobre cómo realizarla. Algunos autores proponen llevarla a cabo mediante hepatectomías parciales, argumentando una mayor seguridad38, mientras que otros (entre los que nos incluimos) creemos que puede suponer un mayor riesgo de hemorragia39. Cada vez son más los autores que defienden que el TOH se debe realizar precozmente (y de una manera especial en mujeres), antes que otras intervenciones invasivas que dificulten técnicamente el posterior TOH, para mejorar los resultados y la supervivencia de los pacientes.
Los pacientes con PQH deben ser priorizados en la lista de espera, ya que cuando el TOH se realiza tardíamente, con una malnutrición manifiesta y con síndrome ocupacional severo, los resultados son claramente peores18,40. En nuestro centro se ha priorizado a estos pacientes mediante la aportación adicional de puntos en el momento de inclusión en lista, al considerarse la PQH una excepción para la aplicación estricta del MELD41.
ConclusiónEl TOH es el tratamiento de elección para la PQH terminal, que presenta una excelente tasa de supervivencia, mayor que en los TOH por otras causas. Estos resultados son mejores cuanto más precozmente se realiza el trasplante, por lo que estos pacientes deberían recibir puntos adicionales para poder utilizar el MELD como sistema válido de priorización en lista.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Parte de la información del manuscrito fue presentada previamente en el XXIII Congreso de la Sociedad Española de Trasplante Hepático, celebrado en Bilbao en octubre de 2011.

