





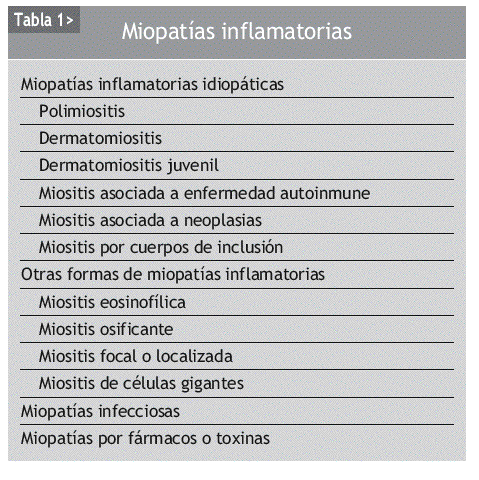
INTRODUCCIÓN
La dermatomiositis (DM) y la polimiositis (PM) son enfermedades inflamatorias autoinmunes de causa desconocida cuya principal diana es el músculo estriado; tienen mecanismos fisiopatológicos distintos y pueden aparecer de forma aislada o bien asociadas a una neoplasia o a una conectivopatía, que, junto con la miositis por cuerpos de inclusión (MCI), se han agrupado bajo el término de miopatías inflamatorias idiopáticas (MII)1. La inflamación del músculo esquelético se suele acompañar de debilidad muscular, elevación de las enzimas musculares y evidencia electrofisiológica e histopatológica de lesión muscular (tablas 1 y 2). Las MII presentan características inmunopatológicas y clínicas heterogéneas, y se caracterizan por la presencia de una lesión inflamatoria muscular asociada a necrosis de células musculares, lo cual se traduce, en la mayoría de los casos, en debilidad muscular. La DM y la PM son consideradas por algunos autores como formas de expresión diferentes dentro del espectro de un mismo proceso patológico, diferenciándose por la asociación o no a manifestaciones cutáneas. No obstante, se han demostrado diferencias significativas en los cambios anatomopatológicos y en el fenotipo del infiltrado inflamatorio de las lesiones musculares de ambos procesos, que sugieren diferentes mecanismos patogénicos entre ellos2.

Se pueden manifestar a cualquier edad, pero se ha observado la existencia de 2 picos de máxima incidencia: uno en la infancia (generalmente < 10 años) y otro entre los 45 y los 60 años. Es 2 veces más frecuente en mujeres que en varones, y en estos últimos la media de edad de inicio es mayor que en las mujeres.
La incidencia de DM y de PM varía en diferentes estudios, y oscila entre 1 y 30 casos por 106 habitantes/año, con una prevalencia de aproximadamente 10 casos por cada 106 habitantes3. En los casos de DM juvenil y en los pacientes con anticuerpos anti-Jo-1 se ha observado una mayor incidencia en los meses de primavera que en el resto del año. En la DM juvenil existen evidencias de una relación con la infección por el virus coxsackie B, que también es más prevalente en los meses de primavera.
ETIOPATOGENIA
La etiología de las miopatías inflamatorias continúa siendo desconocida. La implicación de los fenómenos de autoinmunidad en su patogenia es sugerida por la posible asociación a procesos autoinmunes o infecciones virales, la evidencia de una miocitotoxicidad mediada por linfocitos T (en el caso de la PM) y de una microangiopatía mediada por el complemento (en la DM)4-6, la presencia en el suero de los pacientes de autoanticuerpos (tabla 3) y la respuesta al tratamiento con corticoides e inmunosupresores.

No está claro el papel que podrían desempeñar los factores genéticos, como ocurre con el polimorfismo HLA-DRB1*0301 para la PM y la MCI7-8, el HLA-DQA1*0501 para la DM juvenil9 o el TNFa-308A para fotosensibilidad en la DM10.
En la DM se observa un infiltrado inflamatorio endomisial centrado en los vasos sanguíneos intramusculares. Este infiltrado presenta un alto porcentaje de linfocitos B y un índice CD4/CD8 elevado. Hay una estrecha relación entre los linfocitos CD4+, los linfocitos B y los macrófagos. Todo ello sugiere la participación en la génesis del proceso de un mecanismo inmunológico mediado por anticuerpos1-2 dirigidos contra la microvascularización intramuscular. Esto justificaría la asociación a múltiples autoanticuerpos cuyo papel patogénico todavía no se ha aclarado.
La observación de que uno de los hallazgos más tempranos y específicos de las biopsias musculares de DM es la presencia de depósitos del complejo de ataque de membrana del complemento (C5b-9) en los capilares sugiere que los anticuerpos dirigidos contra la microvascularización desencadenarían la activación local del complemento1-5. Depósitos similares de C5b-9 se han demostrado en la microvascularización dérmica de estos pacientes6. Una vez activado el complemento por dichos autoanticuerpos y depositado el C5b-9 en la microvascularización, se produciría una progresiva necrosis vascular con reducción del número de capilares por fibra. Esto conduciría a una isquemia con destrucción de fibras e inflamación secundaria. Finalmente se produciría la característica atrofia perifascicular.
ANATOMÍA PATOLÓGICA
La biopsia muscular es la prueba para establecer el diagnóstico definitivo de las MII y para excluir otras enfermedades neuromusculares. Tanto la DM como la PM se caracterizan por manifestaciones o cambios histológicos característicos que resultan de la combinación de 3 procesos histopatológicos básicos: necrosis segmentaria de fibras musculares, inflamación intersticial y vasculopatía.
Para que la biopsia muscular sea diagnóstica es fundamental seleccionar para dicha exploración un músculo afectado y que no haya sido utilizado para la práctica de electromiografías. A pesar de que la musculatura de las cinturas escapular y pélvica es la más frecuentemente afectada por el proceso, a veces puede ser difícil identificar los grupos musculares involucrados. Algunos autores recomiendan biopsiar 2 grupos musculares para aumentar la rentabilidad diagnóstica y, si es posible, guiados por imagen de resonancia magnética (RM). En dos tercios de las biopsias realizadas los hallazgos son diagnósticos, y las biopsias musculares son normales hasta en el 10% de los casos.
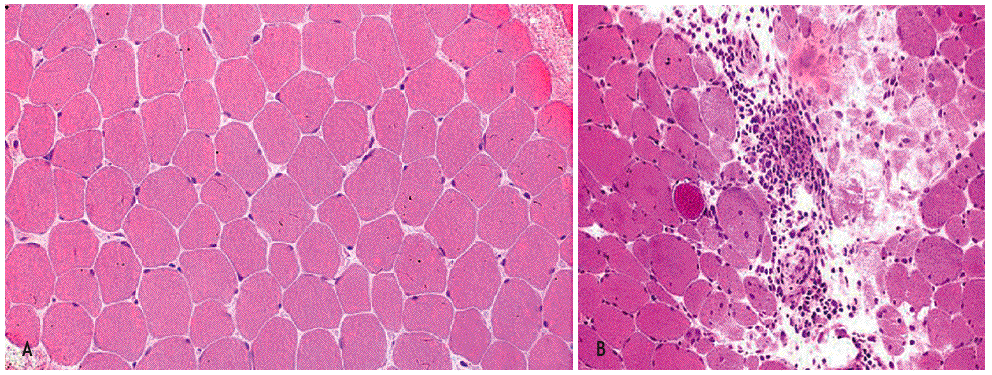
La biopsia muscular (fig. 1) de la polidermatomiositis presenta una reacción inflamatoria celular de intensidad variable y constituida fundamentalmente por linfocitos B y T activados, células natural killer (NK) y macrófagos2,11. La distribución del infiltrado varía de uno a otro proceso: puede ser perimisial o endomisial y se distribuye preferentemente en las regiones perinecróticas o perivasculares12,13. Ambos procesos se diferencian también por el fenotipo celular de infiltrado inflamatorio y por la presencia o no de lesión endotelial. En la DM, el infiltrado inflamatorio es de predominio endomisial y perivascular; se observan necrosis de fibras musculares y atrofia perifascicular, así como lesión y necrosis endotelial de los capilares intramusculares. El infiltrado inflamatorio perivascular está constituido fundamentalmente por macrófagos, linfocitos B y células T CD4+14. Las células endoteliales lesionadas presentan estructuras tubulorreticulares intracitoplasmáticas y existen depósitos vasculares de inmunoglobulinas (IgG, IgM), de factor C3 del complemento y del complejo de ataque de membrana del complemento (C5b-9)4,15. Esto se asocia a fenómenos de trombosis de arterio las y necrosis venular con disminución de la red capilar4.
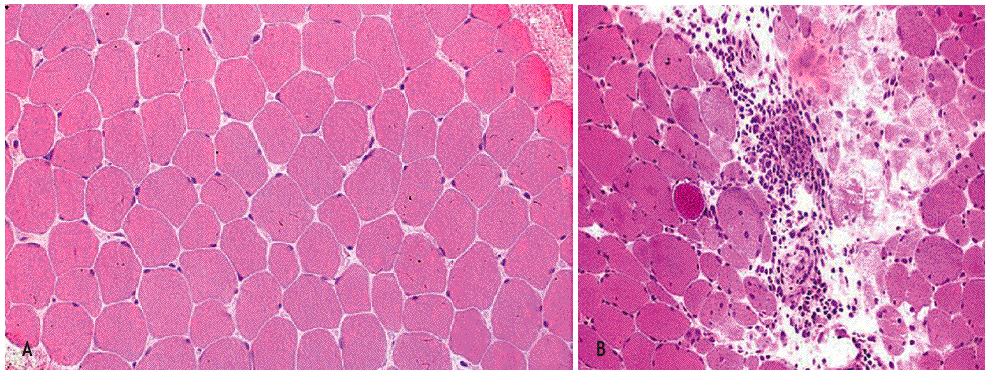
Figura 1 Imagen histológica de un músculo normal y de una miopatía inflamatoria idiopática (MII). En A se ilustra un músculo normal. En B se observa la anatomía patológica típica de la dermatomiositis con un infiltrado inflamatorio de predominio endomisial y perivascular, con necrosis de fibras musculares y atrofia perifascicular, así como lesión y necrosis endotelial de los capilares intramusculares. (De Miller ML, Up To Date.)
En la PM la necrosis de fibras musculares es difusa, con infiltrados inflamatorios perinecróticos y perimisiales. La lesión no está centrada por vasos sanguíneos, y no se observa lesión vascular ni formaciones tubulorreticulares endoteliales. Tampoco se objetivan atrofia perifascicular ni depósitos vasculares de inmunoglobulinas o complemento. El infiltrado inflamatorio está constituido fundamentalmente por macrófagos, linfocitos CD8+ citotóxicos y células NK, con escasez de linfocitos B y de CD4+.
En las MII existe además un patrón degenerativo con presencia de vacuolas y una acumulación de proteínas o de sustancia amiloide que podría actuar de forma independiente.
Las alteraciones histológicas de las biopsias de lesiones cutáneas de DM dependen del estadio evolutivo de la enfermedad. En las lesiones agudas, los cambios histológicos recuerdan a los propios del lupus eritematoso subagudo. En lesiones más avanzadas los hallazgos histológicos son similares a los de la esclerodermia. Los estudios inmunohistoquímicos demuestran la ausencia de fluorescencia en la unión dermoepidérmica, si bien se pueden observar depósitos globulares de IgG, IgM y ocasionalmente de IgA en la dermis superficial. Hay depósitos de C5b-9 en las paredes vasculares y en la unión dermoepidérmica6.
MANIFESTACIONES CLÍNICAS
Manifestaciones musculares
De forma insidiosa (semanas a meses), en la DM y la PM aparece una debilidad muscular proximal y simétrica, y menos frecuentemente mialgias. El paciente puede referir dificultad al subir escaleras, levantarse de una silla, peinarse o vestirse. La debilidad se puede extender a los músculos del cuello, dificultando la posición erecta de la cabeza, y a la musculatura faríngea y del tercio superior del esófago, provocando disfagia. En la exploración física no se detecta debilidad muscular distal, dolor significativo a la palpación muscular, fasciculaciones o anomalías en la musculatura facial o extraocular.
La MCI afecta de forma asimétrica a la musculatura tanto proximal como distal, especialmente al cuádriceps y a los flexores de la muñeca, de los dedos y del tobillo, lo que provoca caídas frecuentes. Es más común en varones (60-70%) de más de 50 años, y su desarrollo y progresión son más lentos (meses a años) que en el resto de MII.
Manifestaciones cutáneas
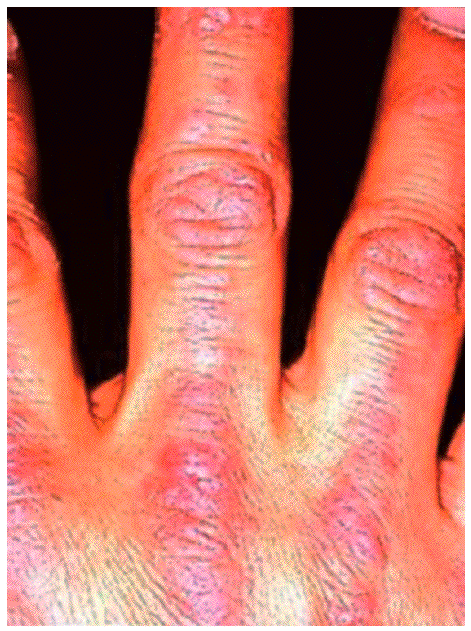
Aparecen en la DM y preceden o acompañan a las manifestaciones musculares, ocasionalmente hasta en varios años. Existen 2 lesiones patognomónicas de DM: a) el eritema en heliotropo (coloración violácea de párpados superiores, asociada muchas veces a edema, presente en menos del 50% de los pacientes) y b) las pápulas de Gottron (lesiones papuloeritematosas descamativas sobre la piel de las articulaciones metacarpofalángicas e interfalángicas, que aparecen en el 60-80% de los casos) (fig. 2). También se observan frecuentemente otro tipo de lesiones cutáneas no específicas, como las erupciones en áreas fotoexpuestas (cuero cabelludo, cara, cuello, espalda, tórax anterior, hombros), que, a diferencia del lupus eritematoso sistémico, pueden ser intensamente pruriginosas. Son también comunes el eritema y las anomalías capilares periungueales (de forma similar a la esclerodermia), la hipertrofia cuticular o las fisuras palmares o digitales ("manos de mecánico"). Especialmente en las formas juveniles de DM pueden desarrollarse calcificaciones subcutáneas.
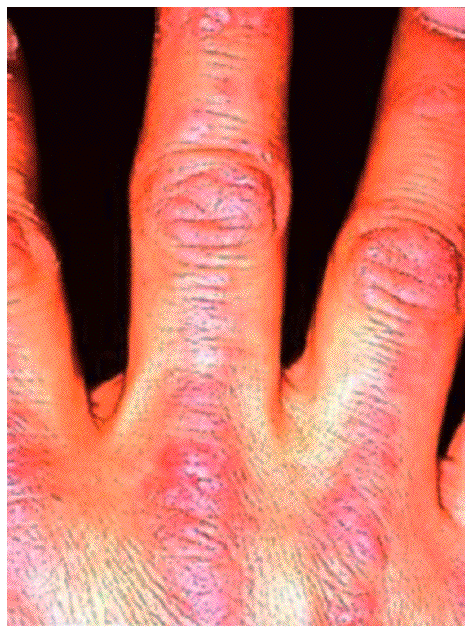
Figura 2 Pápulas de Gottron. (De Miller ML, Up To Date.)
Dermatomiositis amiopática
La DM amiopática o DM sine miositis es una variante en la que los pacientes tienen manifestaciones cutáneas sin evidencia clínica o de laboratorio de afectación muscular durante más de 6 meses (algunos de estos sujetos pueden desarrollar síntomas musculares manifiestos al cabo de años). Este término excluye a los pacientes con DM y afectación muscular subclínica en los que las pruebas complementarias enzimas musculares, electromiograma (EMG), RM o biopsia muscular demuestran algún grado de inflamación muscular (DM hipomiopática). A veces la erupción cutánea característica de la DM es transitoria o no es reconocida (p. ej., individuos de raza negra) y se emite un diagnóstico erróneo de PM. Estos casos (DM sine dermatitis) se identifican correctamente con la biopsia muscular.
Otras manifestaciones
La enfermedad pulmonar intersticial puede aparecer en más del 30% de sujetos con DM o PM y se relaciona con una mayor morbimortalidad. Incluso en sujetos con DM amiopática se ha descrito una incidencia del 13% de enfermedad pulmonar intersticial. Los anticuerpos contra la enzima histidil ARNt sintetasa (anti-Jo-1) se detectan en menos de la mitad de estos pacientes, y hasta en una tercera parte de los casos la afectación pulmonar precede al desarrollo de los síntomas musculares. Otra potencial complicación pulmonar es la neumonía por aspiración, favorecida por la disfagia propia de la miopatía. La afectación cardíaca, en contra de lo esperado, es poco frecuente, y se han descrito diversos tipos de arritmias.
Cuando la PM o la DM están asociadas a otra conectivopatía pueden existir síntomas generales (astenia, anorexia, fiebre, pérdida de peso), artralgias o artritis y fenómeno de Raynaud.
Neoplasias asociadas
Los pacientes con DM tienen un riesgo de 3 a 6 veces mayor que la población general de desarrollar una neoplasia, especialmente a partir de los 60 años de edad, aunque puede asociarse a un tumor en pacientes más jóvenes. Para la PM y la MCI posiblemente ese riesgo sea ligeramente superior al de la población general. Los tipos de tumores más comunes son los de ovario, tracto digestivo, pulmón, mama y linfomas no Hodgkin. Es necesario efectuar un cribado de neoplasia en todo adulto de más de 50 años diagnosticado de MII.
CLASIFICACIÓN
Ha habido múltiples clasificaciones de las MII16-18. El primer intento de llevar a cabo su clasificación integrada fue realizado por Bohan y Peter19. Algunas deficiencias de su clasificación han llevado a plantear modificaciones que han permitido establecer otras clasificaciones, como la que considera la existencia de las siguientes categorías: a) PM; b) DM; c) miositis asociada a neoplasia; d) DM/PM infantil; e) miositis asociada a otras conectivopa-tías (síndromes de solapamiento); f) DM amiopática, y g) MCI20.
Bohan y Peter establecieron los criterios clasificatorios de la PM y la DM (tabla 2): a) debilidad simétrica de los músculos de las cinturas escapulares y pélvica, de los músculos flexores del cuello, progresiva en el curso de semanas o meses, con o sin disfagia o afectación de la musculatura respiratoria; b) biopsia muscular compatible; c) elevación plasmática de enzimas musculares, especialmente de la creatinfosfocinasa y frecuentemente de la aldolasa, la aspartato aminotransferasa, la alanina aminotransferasa y la lactato deshidrogenasa; d) tríada electromiográfica de unidades motoras polifásicas pequeñas y cortas, fibrilaciones, ondas positivas agudas e irritabilidad insercional y descargas repetitivas anómalas de alta frecuencia, y e) cualquiera de las manifestaciones cutáneas características de la DM.
Los pacientes que cumplan los 4 criterios musculares tendrán un diagnóstico definitivo de PM. El cumplimiento de 3 criterios musculares permite establecer el diagnóstico de probabilidad, y el de posibilidad cuando sólo se cumplan 2.
En los pacientes con 3 o más criterios musculares y el criterio de afectación cutánea se establecía el diagnóstico de certeza de DM; el de probabilidad con 2 criterios musculares y el criterio de afectación cutánea, y el de posibilidad con un criterio muscular y el criterio cutáneo.
Recientemente se ha sugerido utilizar la inmunohistoquímica para identificar el complejo principal de histocompatibilidad clase I y el marcador celular CD8 en las muestras de biopsia muscular, que podrían distinguir las PM y las MCI de otras miopatías20.
PRONÓSTICO
Con los tratamientos actuales se consiguen supervivencias superiores al 85%, si bien hasta en la mitad de los casos persiste cierto grado de debilidad y cerca del 20% de los pacientes quedan significativamente limitados, especialmente los niños, con contracturas residuales y calcinosis. Aproximadamente la mitad de los pacientes se recuperan y pueden suspender el tratamiento tras 5 años de iniciarse el proceso, mientras que el 20% requerirán continuarlo por la persistencia de actividad de la enfermedad.
El índice de mortalidad en pacientes con DM y PM es, aproximadamente, 4 veces superior al de la población general. Las principales causas de muerte son las infecciones respiratorias, generalmente secundarias a aspiraciones, la insuficiencia cardíaca, la malnutrición secundaria a la disfagia intensa, la debilidad, las neoplasias asociadas y los efectos secundarios del tratamiento. Por ello el pronóstico empeora en los pacientes con afectaciones viscerales, especialmente pulmonares, cardíacas y renales. La asociación a ciertos autoanticuerpos que determinan la posibilidad de afectación visceral, como los anti-Jo-1 y PL 12, puede tener significado pronóstico. También se relaciona con un peor pronóstico el hecho de pertenecer al sexo femenino, ser de raza negra, edades de inicio avanzadas, presentar formas clínicas con afectación importante desde la presentación inicial o inicio muy tardío del tratamiento. Son indicadores de mal pronóstico la presencia de disfagia o la asociación con cáncer. El pronóstico no varía entre las formas clásicas y amiopática de DM. Las formas asociadas a cáncer pueden mejorar con el tratamiento de éste.
A pesar de todos estos datos, no hay un patrón de comportamiento claramente definido que nos ayude a determinar el curso que va a seguir la enfermedad en un paciente en concreto. En cualquier momento pueden producirse recaídas, y éstas tienen peor pronóstico si se dan tras reducciones excesivamente rápidas del tratamiento corticoide.
También es importante el inicio precoz de actividad física tras la mejoría de los signos inflamatorios musculares, puesto que reduce considerablemente el riesgo de desarrollar miopatía por desuso, que es una de las principales secuelas de la enfermedad.
TRATAMIENTO
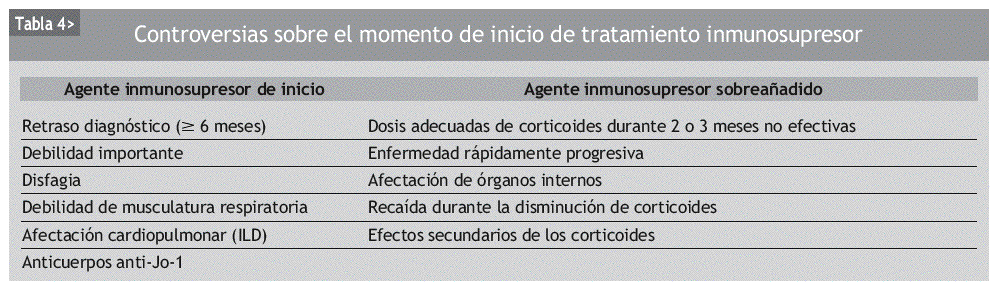
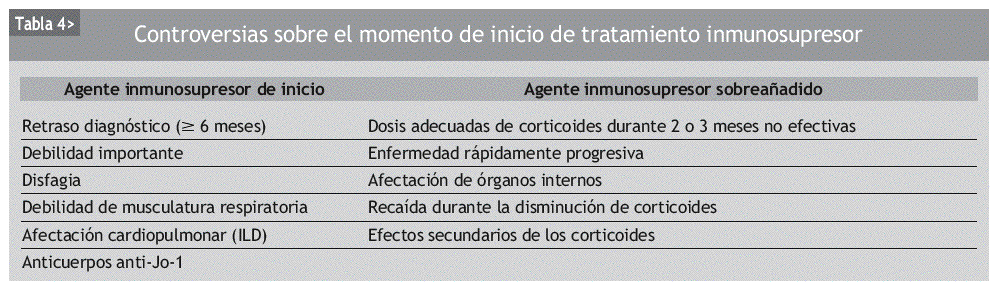
El abordaje del tratamiento es principalmente empírico y no hay acuerdo sobre cuál es el manejo más óptimo de las miositis. Además, actualmente tampoco hay consenso a la hora de iniciar un tratamiento con inmunosupresores, aunque la tendencia sea añadirlos lo más pronto posible (tabla 4).
En las enfermedades poco prevalentes es un auténtico reto poder reunir pacientes para ensayos terapéuticos a gran escala. Por este motivo, lo que suele ocurrir es que se utilizan fármacos que funcionan bien en otras enfermedades inflamatorias sistémicas, especialmente si en la investigación básica hay datos que apoyen su eficacia en la supresión de la inflamación.
Sin una terapia estándar definida y sin instrumentos consistentes de mejoría es imposible definir si un paciente es resistente al tratamiento instaurado. El IMACS (Internacional Myositis Assessment and Clinical Studies group)21 ha desarrollado una serie de parámetros y de definiciones de mejoría clínica para estudios de las miositis. Una vez que se generalice el uso de dichos parámetros en los ensayos clínicos seremos capaces de entender mejor los efectos de los tratamientos y de poder realizar comparaciones entre los distintos fármacos22.
Glucocorticoides
Los glucocorticoides sistémicos son el tratamiento empírico de elección. A falta de estudios doble ciego, estos fármacos son claramente eficaces y se acepta que disminuyen la morbilidad y la mortalidad en las formas tanto juveniles como del adulto. El inicio precoz del tratamiento permite disminuir las dosis totales y el tiempo de duración del tratamiento. Los glucocorticoides no son útiles para el tratamiento de las contracturas residuales, y se supone que su acción consiste en mejorar la vasculopatía y disminuir el depósito de los componentes finales del complemento.
El tipo de enfermedad puede influir en la respuesta al tratamiento. Así, por ejemplo, se ha descrito una peor respuesta en los casos de síndrome de solapamiento que en las formas puras de DM. Hay autores que consideran que es esencial iniciar el tratamiento de la enfermedad con dosis elevadas de glucocorticoides, ya que con ello se consigue una mayor efectividad y se reducen las secuelas del proceso. Se entiende por dosis elevadas las que corresponden a 1-2 mg/kg/día de prednisona y se mantienen entre 3 y 4 semanas. Las dosis inferiores, aparte de no ser efectivas, suponen un mayor riesgo de desarrollo de efectos secundarios derivados de la corticoterapia, al alargar la duración total del tratamiento.
Una vez solucionada la fase aguda del proceso, las dosis de glucocorticoides se deben reducir progresivamente en los meses siguientes. Se considera necesaria una reducción gradual y lenta de las dosis diarias que permita una normalización de las concentraciones séricas de las enzimas musculares y de la fuerza muscular. Se recomiendan disminuciones de dosis de entre 5 y 10 mg/día cada 3-4 semanas. Las dosis se reducen hasta alcanzar las mínimas necesarias para mantener el proceso bajo control. Habitualmente se llega a conseguir mantener controlado al paciente con dosis de entre 10 a 25 mg a días alternos o las dosis equivalentes diarias.
Algunos pacientes no responden al tratamiento con prednisona y se vuelven corticorresistentes. Se consideran como tales los pacientes que, tras 2-4 meses de tratamiento a dosis de entre 40 y 80 mg/día, no presentan respuesta favorable para la mejoría de la fuerza muscular, de la sensación de bienestar o la normalización de las enzimas musculares. Dado que estas últimas pueden normalizarse pese a la persistencia de la actividad del proceso, actualmente se tiende a ajustar el tratamiento en función del grado de recuperación de la fuerza muscular. El problema que se plantea en el ajuste de las dosis es la posibilidad de que un paciente desarrolle una miopatía esteroidea, con lo cual la clínica muscular puede persistir o empeorar pese a la normalización de las enzimas musculares. Esta situación plantea la duda acerca de si dicho empeoramiento es secundario a una reagudización del proceso, con lo cual el paciente requeriría un aumento en las dosis de glucocorticoides, o de la citada miopatía esteroidea, en cuyo caso se precisa reducir las dosis. La tendencia a la normalización de los parámetros enzimáticos orienta hacia la segunda, por lo que se recomienda reducir las dosis de glucocorticoides y adoptar una actitud expectante sobre la evolución de la clínica muscular. Ni la repetición de la biopsia muscular ni la práctica de un nuevo electromiograma parecen ser útiles en la diferenciación de estas 2 situaciones. Se están investigando una serie de marcadores que se relacionan con el grado de lesión de la célula endotelial, así como la posible utilidad de técnicas de imagen como posibles parámetros de valoración de la actividad clínica con el fin de evitar los problemas en el ajuste de los tratamientos.
La utilización de metilprednisolona en bolo intravenoso (máximo de 1 g/día) se puede aplicar como tratamiento inicial en pacientes con formas graves de la enfermedad o en casos resistentes al tratamiento oral, especialmente en la DM juvenil, aunque esta recomendación se sustenta únicamente en series de casos publicados23-25.
Inmunosupresores
Si se presenta una corticorresistencia (hasta en el 20% de los pacientes), se plantea la necesidad de recurrir a otros tratamientos. Además de ésta, hay más situaciones en las que se plantea la necesidad de utilizar otros inmunosupresores:
La necesidad de reducir las dosis de glucocorticoides, pese a una buena respuesta, a fin de reducir los efectos secundarios derivados de su uso.
Cuando los repetidos intentos de reducción de dosis de glucocorticoides provocan reagudizaciones del proceso.
Cuando el paciente presenta una enfermedad rápidamente progresiva con debilidad intensa e insuficiencia respiratoria.
En una revisión de la Cochrane Database, Choy et al26 examinaron la eficacia de los agentes inmusosupresores y sólo identificaron 6 ensayos clínicos aleatorizados potencialmente relevantes. La azatioprina y el metotrexato son los agentes más comunes añadidos a la prednisona. Un ensayo de azatioprina frente a placebo27 mostraba mejoría a los 3 meses en el grupo tratado, pero la mejoría comparada con los controles no era significativa. En una extensión de dicho estudio sí alcanzó diferencia significativa28. En un estudio de Villalba et al29 el metotrexato intravenoso con ácido folínico fue útil en algunos pacientes resistentes al tratamiento, y la combinación de azatioprina oral diaria junto a metotrexato semanal mostraba una tendencia a mayor mejoría.
La preferencia por uno u otro fármaco se basa en la experiencia personal y la relación beneficio/seguridad de los mismos.
Azatioprina
Se administra oralmente, en dosis de entre 1 y 3 mg/kg/día. Para muchos autores es el fármaco preferido dada su eficacia, la tolerancia por parte del paciente y su relativa seguridad. Requiere de 3 a 6 meses de mantenimiento para producir sus efectos, por lo cual deberá mantenerse como mínimo ese período antes de considerarse no efectivo.
Metotrexato
Se administra por vía oral o parenteral, a una dosis de 15-25 mg semanales. Actualmente está desplazando a la azatioprina como primer inmunosupresor empleado. Su efecto se instaura antes que en el caso de la azatioprina, y la dosis semanal es muy cómoda.
Hidroxicloroquina
A menudo se utiliza para el tratamiento de las manifestaciones cutáneas de la DM, a una dosis de hasta 200 mg/12 h.
Ciclofosfamida
Administrada por vía intravenosa u oral (2-2,5 mg/kg), los resultados referentes a su efectividad son contradictorios. Parece estar indicada en pacientes con enfermedad pulmonar intersticial o en casos resistentes a otros inmunosupresores, incluyendo inmunoglobulinas. Su uso estaría también justificado en DM con vasculitis, afectación de la musculatura respiratoria y/o bulbar. En un estudio en 12 casos de DM juvenil tratados con bolos intravenosos a una dosis de 0,5-1 g/m2, 2 fallecieron precozmente por complicaciones (eran pacientes con ventilación asistida por distrés respiratorio) y los 10 restantes mostraban mejoría de la fuerza muscular, actividad de la enfermedad extramuscular y marcadores bioquímicos30.
Ciclosporina A (CSA)
Este fármaco supresor de la función de las células T se ha utilizado con limitado éxito; se han descrito resultados favorables en el tratamiento de casos infantiles de DM31. En un ensayo de 20 pacientes con DM y 16 con PM aleatorizado frente a metotrexato32 se obtuvo una mejoría similar con ambos fármacos, aunque con la CSA la mejoría se alcanzó más tarde (a pesar de un descenso más rápido de los valores séricos de IL-1Ra). En el caso particular de la asociación de estas patologías con enfermedad intersticial pulmonar, las series de casos publicadas muestran resultados dispares. En general, mejora la resdillo> Tacrolimus
Es un agente similar a la CSA. En un estudio en 8 pacientes35 con miositis resistente a corticoide, 6 de ellos con anti-Jo-1 positivo, se utilizó a una dosis de 0,075 mg/kg/día en 2 tomas diarias y todos los pacientes mejoraron, aunque presentaron una recaída al bajar la dosis o suspender el tratamiento. El tratamiento tópico para las lesiones cutáneas en un estudio en 5 pacientes36 no aportó diferencias frente a la actitud de no tratar.
Micofenolato de mofetilo
El ácido micofenólico, su metabolito activo tras hidrólisis hepática, es un inhibidor de la proliferación de los linfocitos T y B. En una serie de 12 pacientes37 con lesiones cutáneas recalcitrantes de DM, 10 presentaron mejoría de los síntomas cutáneos y musculares tras 4 a 8 semanas del inicio del tratamiento a una dosis de 2-3 g/día, y los efectos secundarios observados fueron leucopenia, trombopenia y elevación de transaminasas. Un paciente desarrolló un linfoma de células B de sistema nervioso central relacionado con el virus de Epstein-Barr.
Otra serie de 7 pacientes con miositis, 4 con DM y 3 con PM, que no habían respondido a otros inmunosupresores, obtuvo respuesta en 6 pacientes38.
Inmunoglobulinas intravenosas (Ig i.v.)
Es un tratamiento con buenos resultados pero con un coste económico elevado. Se purifican a partir de plasma humano obtenido de miles de donantes. El mecanismo de acción podría ser el bloqueo de los receptores Fc, la unión a componentes del complemento y su inhibición, la neutralización directa de autoanticuerpos por antiidiotipos o la reducción de la vida media de autoanticuerpos por saturación del receptor de rescate de la IgG FcRn.
Uno de los pocos ensayos clínicos39 frente a place bo en el campo de las MII ha demostrado que es un tratamiento eficaz en la DM. En este ensayo se aleatorizaron 15 pacientes para recibir placebo o Ig i.v. 1 g/kg/día, 2 días cada mes durante 3 meses, con la posibilidad de cruce 3 meses más. Los 8 pacientes que recibieron Ig i.v. inicialmente tenían una mejoría significativa de la fuerza con respecto al grupo placebo, y 9 de 12 tras el cruce, además de 2 pacientes con moderada mejoría. Ninguno de los 11 pacientes y de la erupción durante el tratamiento con placebo obtuvo una mejoría significativa. Cherin et al40 describen un estudio abierto en 35 pacientes con PM que fallaron al tratamiento con esteroides orales y al menos a un inmunosupresor. El 71% (25/35) obtuvo una mejoría significativa de la fuerza muscular a los 3 meses del tratamiento, con reducciones significativas de la creatincinasa. Los síntomas esofágicos desaparecieron en 8 de 11 pacientes. Las dosis de esteroides también se pudieron reducir.
Rituximab
Anticuerpo monoclonal quimérico frente a linfocitos B con resultados prometedores, como se observa en un estudio piloto abierto41 en el que 5 pacientes con DM refractaria al menos a 3 tratamientos y 1 paciente de reciente diagnóstico reciben 4 dosis de 375 mg/m2 con mejoría sustancial del eritema y de la fuerza muscular. Tres pacientes con afectación pulmonar obtuvieron mejoría.
Otras series o casos aislados demostraron la utilidad de rituximab en la PM y en el síndrome antisintetasa42-44.
Etanercept
Se utilizó junto a inmunosupresores orales en un estudio que recogía a 6 pacientes45. Todos presentaron exacerbaciones y tuvieron que seguir con metotrexato, azatioprina o CSA. No tuvo capacidad para disminuir los corticoides, y no hubo cambios en la electromiografía ni descenso de las enzimas musculares.
Infliximab
Series como la de Labioche et al46 demostraron sólo mejorías parciales incluso en respondedores y utilizando dosis más elevadas que las típicamente empleadas para la artritis reumatoide, lo que conlleva además un aumento del riesgo de infecciones.
Clorambucilo
Este agente alquilante ha sido útil en asociación con metotrexato y prednisona.
Fludarabina
Es un análogo de la adenosina que se ha utilizado con escasa respuesta en un estudio piloto en 16 pacientes con DM y PM47.
Eculizumab
Es un anticuerpo monoclonal frente a C5. En un estudio en 10 pacientes se obtuvo mejoría de las lesiones cutáneas frente a placebo48.
Otros procedimientos
Se han ensayado procedimientos como el recambio plasmático, cuyos resultados son contradictorios en las diferentes series, y la irradiación corporal total, que se ha mostrado útil en casos desesperados; se consiguieron remisiones prolongadas, pero sus efectos secundarios obligan a considerar seriamente la necesidad de practicarlo. Finalmente, se ha descrito la práctica de timectomías o la utilización de mercaptopurina en algunas series.
RESUMEN
Los corticoides siguen siendo la primera línea de tratamiento.
Los bolos de metilprednisolona son útiles en las manifestaciones clínicas graves.
Los inmunosupresores se emplean como terapia de inicio junto a los esteroides o:
* Si hay resistencia a los corticoides.
* En la enfermedad rápidamente progresiva.
* En la afectación de órganos internos.
* En secundarismos del tratamiento con corti coides.
El metotrexato es efectivo y es el primer fármaco inmunosupresor que debe utilizarse.
La azatioprina también puede emplearse como primer inmunosupresor.
En las MCI ningún tratamiento suele mejorar al paciente.
La ciclosporina A puede utilizarse sola o en combinación con metotrexato con buenos resultados, especialmente en caso de enfermedad pulmonar intersticial.
Las inmunoglobulinas se consideran en casos refractarios y en la disfagia.
Hay que valorar el rituximab por remisiones más largas y menor coste.
La ciclofosfamida se usa en casos refractarios con vasculitis, neumonitis o afectación de la musculatura respiratoria.
El tacrolimus y el micofenolato han demostrado buenos resultados y se esperan estudios aleatori-zados.
AGRADECIMIENTOS
Deseamos expresar nuestro agradecimiento a la Sociedad Española de Reumatología por la invitación para realizar este trabajo.