




INTRODUCCIÓN
La hepatitis autoinmune (HAI) es una hepatopatía inflamatoria, crónica y progresiva, de etiología desconocida; su desarrollo se atribuye a una compleja interacción entre factores ambientales en pacientes genéticamente predispuestos y a alteraciones en los mecanismos de regulación del sistema inmunológico. A grandes rasgos se caracteriza por la presencia de autoanticuerpos circulantes, hipergammaglobulinemia, cambios histológicos característicos y una buena respuesta al tratamiento inmunosupresor.
EPIDEMIOLOGÍA
La HAI afecta a ambos sexos y a todas las edades pero es mucho más prevalente en la mujer (3,6 veces más frecuente que en el varón) y existe un pico de incidencia alrededor de los 50 y 60 años1. Se describe con mayor frecuencia en la raza blanca y en la población europea. Es frecuente que aparezca asociada a otras enfermedades extrahepáticas autoinmunes y se comporte como un proceso multisistémico. La prevalencia estimada en Europa Occidental y en Norteamérica varía desde 0,4 hasta 17 casos por 100.000 habitantes2,3.
PATOGENIA
Como ya se ha mencionado, la etiología de la HAI es desconocida, y se considera que el desarrollo de la enfermedad es el resultado de la combinación de factores ambientales que desencadenarían el proceso, una susceptibilidad genética del huésped y alteraciones en la regulación del sistema inmunitario.
No se conocen con exactitud los factores desencadenantes, aunque se han incluido agentes infecciosos (virus hepatotropos como el virus de la hepatitis A, B o C, otros virus como el virus del sarampión, citomegalovirus, virus de Epstein-Barr, etc.) así como tóxicos o ciertos fármacos (entre los más conocidos, metildopa, diclofenaco, interferón, atorvastatina, nitrofurantoínas, etc.) y productos de herboristería.
De forma simple y a modo de recordatorio podemos resumir que los antígenos (o en este caso autoantígenos) son procesados y presentados por las células presentadoras de antígenos (CPA), junto a las moléculas HLA tipo II, al linfocito T helper CD4. Éste reconocerá el antígeno a través de su receptor antigénico (TCR) y se activará. A partir de esta primera señal, el linfocito T CD4 activado secretará una serie de citocinas que activarán el resto del sistema inmune.
Con el fin de buscar asociaciones entre determinados genes y su papel en la regulación de la respuesta del sistema inmune, se han estudiado los haplotipos HLA, hallándose una mayor prevalencia de los alelos HLA de clase II DR3 y DR4, en los pacientes afectos de HAI, sobre todo en la HAI tipo I2,4. Es importante remarcar que la susceptibilidad genética no depende de un único gen, sino que depende de la presencia de polimorfismos en varios genes, tanto los correspondientes a la región HLA como otros implicados en la inmunorregulación.
Una vez el factor desencadenante actúa en un huésped genéticamente predispuesto, el mecanismo por el que se desencadena un defecto en la regulación del sistema inmune que conduce a la HAI no está bien establecido. Se han propuesto distintas hipótesis para explicar este hecho, y la más aceptada es la del mimetismo antigénico, según la cual distintos antígenos externos compartirían secuencias de aminoácidos con antígenos propios, lo que provocaría una respuesta inmune errónea. Entre otras hipótesis que se barajan están la formación de superantígenos y la aparición de neoepítopos debido a proteínas que han sido presentadas de manera inadecuada en el timo o han sido modificadas por acción de agentes ambientales.
MANIFESTACIONES CLÍNICAS
La HAI tiene un curso fluctuante y heterogéneo, lo que determina la variabilidad de sus manifestaciones clínicas.
La presentación clínica de la HAI es muy inespecífica. Puede iniciarse con un cuadro clínico insidioso, con malestar general, astenia y anorexia, aunque en un alto porcentaje de casos los pacientes se encuentran asintomáticos y el diagnóstico se basa entonces en los hallazgos bioquímicos. Otros signos que con frecuencia se presentan asociados son el acné y las telangiectasias cutáneas, así como alteraciones en la menstruación e incluso amenorrea. En algunos casos puede presentarse como una hepatitis aguda fulminante, con ictericia y signos de insuficiencia hepatocelular grave.
En aproximadamente el 25% de los casos la enfermedad se presenta de forma aguda, y en una pequeña parte de éstos en forma de fallo hepático fulminante. Frecuentemente estos pacientes presentan largos períodos de enfermedad subclínica antes o después del diagnóstico.
La exploración física también varía según el estadio de la enfermedad. Puede oscilar desde un examen físico normal hasta presentar hepatomegalia, esplenomegalia o estigmas de hepatopatía crónica y/o ictericia. Los pacientes con HAI pueden presentar síntomas asociados relacionados con la presencia de otros trastornos autoinmunes, entre un 10-50% de los casos (tabla 1)5. Las enfermedades más frecuentemente relacionadas son la tiroiditis, la diabetes mellitus tipo 1, la enfermedad celíaca, los trastornos reumatológicos inflamatorios y la colitis ulcerosa (más frecuente en pacientes con colangitis esclerosante primaria).

DIAGNÓSTICO
Para realizar el diagnóstico de HAI se debe excluir la presencia de otras posibles causas de lesión hepática aguda y crónica, como infecciones por virus hepatotropos (VHC, VHB, VHD), hepatopatías por tóxicos (alcohol) y fármacos o por depósito (hemocromatosis, enfermedad de Wilson)6.
Datos de laboratorio
Las características principales que definen a la HAI son títulos altos de autoanticuerpos (>1/40), predominantemente ANA, aSMA, aLKM-1, e hipergammaglobulinemia policlonal, sobre todo a expensas de IgG, generalmente en valores de entre 1,2 hasta 3 veces los normales. Sin embargo, valores normales de IgG no excluyen el diagnóstico de HAI1. Esta hipergammaglobulinemia está asociada generalmente a la presencia de autoanticuerpos circulantes, que son especialmente útiles para realizar el diagnóstico. Por otro lado es relativamente frecuente el déficit de IgA en niños, sobre todo en los que presentan HAI tipo 2.
En el ámbito bioquímico, la elevación de los valores de transaminasas es la alteración más frecuentemente encontrada. Sin embargo, en algunos casos pueden encontrarse datos de colestasis con importante elevación de la bilirrubina conjugada y de la fosfatasa alcalina, y en estos casos debe excluirse la presencia de patología obstructiva de la vía biliar, formas colestásicas de hepatitis virales, cirrosis biliar primaria o colangitis esclerosante primaria.
Es importante destacar que ni las alteraciones analíticas del perfil hepático ni los valores de gammaglobulina son buenos predictores de la lesión histológica hepática ni de la presencia o ausencia de cirrosis6.
Histología
La biopsia hepática es esencial para establecer el diagnóstico y para determinar la gravedad de la enfermedad, así como la necesidad de tratamiento6. No existe un patrón histológico específico en la HAI, y generalmente es superponible al que se encuentra en cualquier otra hepatopatía crónica, por lo que no es suficiente para realizar el diagnóstico de HAI. Los hallazgos de la biopsia incluyen el infiltrado periportal por células mononucleares (hepatitis periportal o de interfase), la infiltración de los espacios porta por células plasmáticas, la frecuente detección de eosinófilos y la disposición de los hepatocitos en rosetas. También se pueden encontrar cambios en los conductos biliares en aproximadamente el 25% de los pacientes con HAI1.
Autoanticuerpos
Los autoanticuerpos anti-nucleares (ANA), anti-músculo liso (SMA) y anti-microsomales hepatorrenales tipo 1 (anti-LKM1) deben ser determinados en todos los pacientes con sospecha clínica, analítica y/o histológica de HAI7. Los ANA son los marcadores tradicionales de la enfermedad y están presentes en el 67% de los casos (en el 13% de forma aislada y en el 54% asociados a SMA). Los SMA están presentes en el 87% de los casos de HAI, bien de forma aislada (33%) o en asociación con los ANA (54%). Los anti-LKM1 se dan típicamente en ausencia de ANA y SMA.
Actualmente existen nuevos anticuerpos que mejoran la precisión diagnóstica y pueden aportar datos pronósticos de la enfermedad6. Generalmente no están disponibles en todos los laboratorios y su valor definitivo está todavía por determinar:
Anticuerpos anti-actina
Tienen mayor especificidad que los SMA para el diagnóstico de HAI. Estudios preliminares han determinado que estos anticuerpos se relacionan con el HLA DR3, una menor edad en el diagnóstico y una peor respuesta al tratamiento con corticoides. Parece que pueden tener cierta utilidad pronóstica para la enfermedad. Sin embargo, presentan una baja sensibilidad para la HAI, lo que limita su utilidad.
Anticuerpos anti-receptor de la asialoglucoproteína (anti-ASGPR)
Pueden coexistir con los ANA, los SMA y los anti-LKM1, y también parece que pueden tener utilidad pronóstica. Su presencia se correlaciona con el grado de actividad histológica, su desaparición con la respuesta al tratamiento y su persistencia con la recidiva tras el tratamiento con corticoides.
Anticuerpos anti-antígenos solubles hepático y hepático-pancreático (anti-SLA/LA)
Son marcadores de alta especificidad (99%) frente a HAI. Se relacionan con formas graves de la enfermedad y con una alta posibilidad de recidiva tras suspender el tratamiento corticoide. Se encuentran en el 10 al 30% de los pacientes con HAI, y en aproximadamente el 10% de los casos son el único anticuerpo que está presente. Tienen una fuerte asociación con HLA DR3 (HLA DRB1*0301).
Anticuerpos anti-antígeno del citosol hepático tipo 1 (anti-LC1)
Raramente se encuentra en pacientes mayores de 40 años, y generalmente se presenta en menores de 20 años. En el 32% de los casos se asocia a anti-LKM1, relacionándose en algunos estudios con la presencia de otros trastornos autoinmunes, ausencia de infección por el virus de la hepatitis C y con una rápida progresión a cirrosis.
Clásicamente la HAI se ha divido en subgrupos en función del patrón serológico que presenta (tabla 2)1,8. De esta forma, puede definirse el tipo I (o tipo lupoide), que corresponde al 80% de los casos, caracterizado por la positividad sérica para ANA, SMA y anticuerpos antiactina. Aunque los anticuerpos SLA/LP son los más específicos para HAI tipo I, éstos sólo se encuentran positivos en un 10 al 30% de los casos. Este tipo aparece en adolescentes y en mujeres con edad posmenopáusica. En las formas juveniles se ha descrito una mayor asociación con el haplotipo HLA-DR3, presentando un curso más agresivo y de difícil control. La forma de inicio a los 50-60 años se ha relacionado con una mayor prevalencia de HLA-DR4, presentando una evolución menos agresiva. La HAI-I también se asocia a otras enfermedades autoinmunes como hipertiroidismo y anemia hemolítica. El tipo II se caracteriza por la presencia de anti-LKM1. Suele afectar a pacientes más jóvenes y también se asocia a otras enfermedades autoinmunes, como diabetes mellitus insulinodependiente, enfermedad tiroidea autoinmune y vitíligo. Su presentación suele ser aguda y fulminante, y generalmente con un peor pronóstico.

CRITERIOS DIAGNÓSTICOS
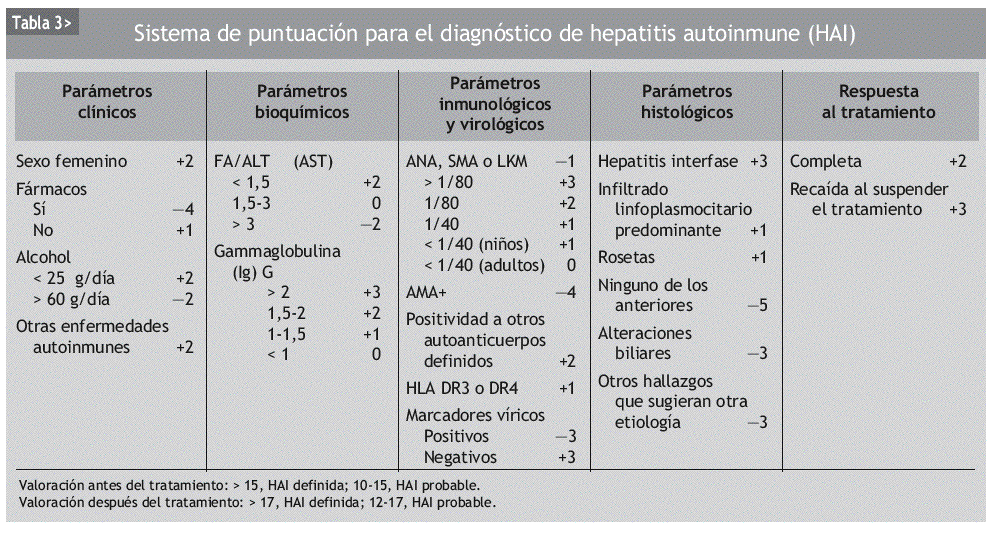
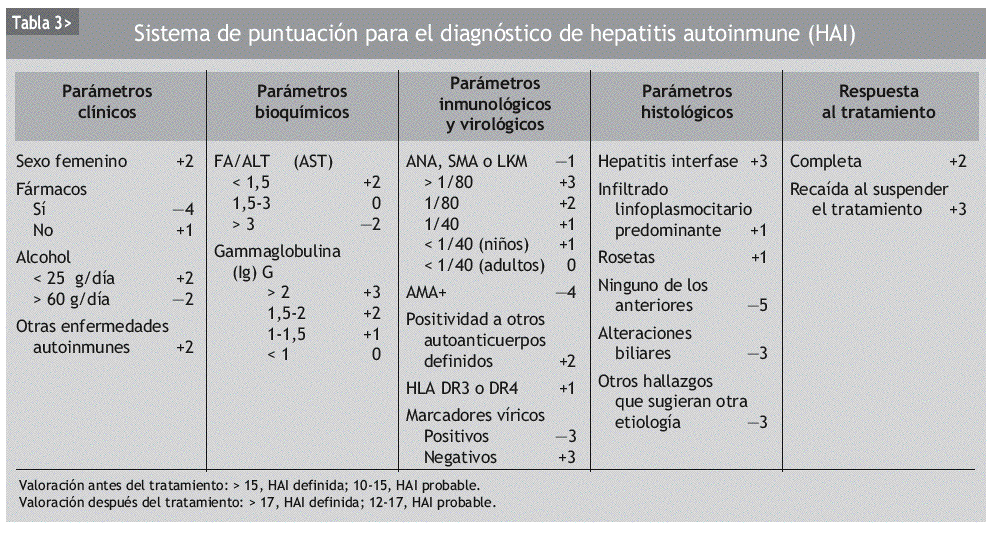
A menudo el diagnóstico puede ser difícil, ya que los criterios mencionados pueden aparecer también en hepatopatías de otros orígenes. Para ayudar a realizar el diagnóstico de HAI, en 1999 un grupo de expertos diseñó un sistema de puntuaciones que evalúa datos clínicos, bioquímicos, serológicos e histológicos, así como la respuesta al tratamiento9. En los pacientes no tratados se considera que el diagnóstico es definitivo si la puntuación es mayor de 15 puntos y probable si es de entre 12 y 17 puntos. En los pacientes que reciben tratamiento inmunosupresor hay certeza diagnóstica si la puntuación es mayor de 17 y probable si varía entre 12 y 17 puntos (tabla 3). Siguiendo este sistema de clasificación, la sensibilidad del diagnóstico es del 97-100% y la especificidad varía entre el 66 y el 92%.
DIAGNÓSTICO DIFERENCIAL
En el diagnóstico diferencial, además de otras hepatopatías no autoinmunes (víricas, tóxica, alcohólica, enfermedad de Wilson, déficit de alfa-1-antitripsina, hemocromatosis), hay que considerar las autoinmunes preferentemente colostásicas (cirrosis biliar primaria, colangitis esclerosante primaria y la mal definida colangitis autoinmune), los síndromes de solapamiento y la hepatitis asociada al lupus sistémico. En cuanto a la hepatopatía asociada al lupus sistémico, se han descrito recientemente anticuerpos antirribosomales P como marcadores de ésta, y no se han detectado en la HAI genuina10.
La distinción entre las entidades anteriores se basa generalmente en datos clínicos, histológicos e inmunológicos. Algunos de los hallazgos que se encuentran en la HAI pueden verse en hepatitis víricas agudas o crónicas, hepatitis tóxicas y otros trastornos hepáticos, lo que dificulta la distinción entre estas entidades.
TRATAMIENTO
La historia natural de la HAI conduce hacia la cirrosis y la insuficiencia hepatocelular. La HAI grave tiene una mortalidad espontánea superior al 90% a los 10 años11. Sin embargo, el tratamiento inmunosupresor provoca la remisión de la enfermedad en el 75% de los casos12,13, disminuye la mortalidad11,14,15 y mejora el pronóstico a corto y a largo plazo. Así, en estudios previos se ha estimado que el tratamiento con prednisona sola o asociada a azatioprina aumenta la supervivencia en el 80 y el 63% a los 2 y 10 años, respectivamente. Sin embargo, el 40% de los enfermos desarrolla una cirrosis a los 10 años de seguimiento a pesar de realizar terapia inmunosupresora16.
En la HAI no grave la expectativa de vida a los 5 años es normal, y en ese tiempo desarrolla cirrosis menos del 17% de los enfermos. En este tipo de HAI se desconoce si el tratamiento inmunosupresor es beneficioso6.
El objetivo del tratamiento es modificar el curso natural de la enfermedad: inducir la remisión y conseguir el mantenimiento de ésta, disminuir el componente inflamatorio y la fibrosis a nivel hepático, mejorar la sintomatología y, por último, disminuir la mortalidad.
Primero de todo es importante definir cuándo se considera una HAI como grave. Básicamente en 3 situaciones6:
Valores de GPT > 10 veces su valor normal.
Valores de GPT > 5 veces y gammaglobulinas > 2 veces su valor normal.
Presencia de necrosis en puentes o multilobu lillar.
El tratamiento inmunosupresor está indicado, según el consenso de la American Association for the Study of Liver Diseases (AASLD) de 20026, en los casos de HAI grave, independientemente de si la enfermedad se encuentra en fase de cirrosis o no. Por lo que se refiere a los casos de HAI no grave, se desconoce si el tratamiento es beneficioso y no existe uniformidad, por lo que debe ser individualizado en pacientes con, al menos, hepatitis de la interfase y/o transaminasas superiores a 2 veces el valor normal. En el embarazo, si se cumplen los criterios de tratamiento, puede instaurarse tratamiento inmunosupresor, ya que es seguro tanto para la madre como para el feto y presenta una respuesta similar a la de la mujer no embarazada.
PAUTAS TERAPÉUTICAS
Diferenciamos una pauta de inducción y una de mantenimiento de la remisión. Para la inducción se aconseja prednisona sola, a dosis de 40-60 mg/día o dosis menores de 30 mg/día, reduciendo también paulatinamente, asociada a azatioprina a dosis de 50 mg/día1,6. Ambas pautas son igual de eficaces y consiguen la remisión en aproximadamente el 75 % de los casos6,17. En la práctica clínica actual, el tratamiento preferido de inicio en adultos con HAI grave es la combinación terapéutica de prednisona con azatioprina, pues el tratamiento combinado produce menos efectos secundarios6. La monoterapia se considera de elección en los casos en que existe citopenia intensa, embarazo, pacientes con patología neoplásica, valores deficitarios de la enzima tiopuril-metil-transferasa eritrocitaria (TPMTe)6.
Una vez conseguida la remisión, debe indicarse el tratamiento de mantenimiento, definido como la dosis mínima eficaz de prednisona, o de prednisona asociada a azatioprina (50-150 mg/día). Esta dosis se obtiene con reducciones progresivas de la medicación de inducción guiadas con determinaciones repetidas de las concentraciones de transaminasas6,17. Otra opción es realizar un tratamiento de mantenimiento sólo con azatioprina a dosis mayores (2 mg/kg/día). En este caso, el tratamiento con prednisona se retirará después de haber pasado al menos 3 meses desde la obtención de la remisión. El mantenimiento con azatioprina en monoterapia es eficaz en el 80% de los casos1,6.
El tratamiento convencional de la HAI debe continuarse en niños y adultos hasta que se consiga la remisión completa y sostenida, el tratamiento fracase, exista una respuesta incompleta o se presente toxicidad de los fármacos. No existe consenso sobre cuánto tiempo debe continuarse el tratamiento de mantenimiento después de conseguir la remisión. Para suspender el tratamiento es indispensable la normalización de las transaminasas, las gammaglobulinas y los valores de IgG, así como la ausencia de actividad inflamatoria en la biopsia hepática. Las concentraciones de anticuerpos no se correlacionan con la actividad de la enfermedad y no es necesario controlarlos para valorar la respuesta al tratamiento. Hay remisión completa de la enfermedad cuando no existe hepatitis de interfase, y para demostrarlo es necesario efectuar un control histológico. Este control debe realizarse preferentemente al menos 1-2 años después del inicio del tratamiento (la remisión histológica no se logra hasta los 6-12 meses del inicio de éste). Si se ha obtenido una remisión completa, se puede intentar retirar el tratamiento si su duración ha sido de 2-3 años, aunque probablemente sea mejor esperar más tiempo, ya que la remisión sostenida tras la retirada del tratamiento es más frecuente si se ha mantenido al menos 4 años18. Según el consenso de la AASLD6, la retirada de la prednisona debe realizarse de forma escalonada en 6 semanas, efectuando controles clínicos y analíticos (transaminasas y gammaglobulina) periódicos. Otra pauta de retirada más lenta consiste en disminuir 2,5 mg de prednisona cada 3 meses y 50 mg de azatioprina cada mes. Una vez retirado el tratamiento se debe revisar a los pacientes periódicamente con controles clínicos y analíticos. Las recidivas suelen ocurrir durante el primer año de haberse suspendido la medicación, pero también es posible que aparezcan después de muchos años.
RECIDIVA
Se estima que en el 50% de los pacientes con HAI aparece recidiva en los 6 meses tras la suspensión del tratamiento, y en el 70 al 80% se produce tras un año de seguimiento sin tratamiento1,6. Se ha sugerido que los factores que pueden predecir la recidiva además de la presencia de cirrosis podrían ser: el tiempo en el que se consigue la remisión bioquímica inicial, los valores elevados de IgG basales y un marcado infiltrado de células plasmáticas portales.
El tratamiento de la recidiva consiste en reintroducir de nuevo el tratamiento de inducción inicial y posteriormente realizar tratamiento de mantenimiento con dosis mínima eficaz de prednisona o bien azatioprina (2 mg/kg/día). A pesar de la recidiva, se debe volver a intentar retirar el tratamiento una vez se cumplan los criterios de remisión completa, dado que el 13% de los pacientes conseguirá mantener la remisión a los 5 años1,6.
FRACASO DEL TRATAMIENTO INMUNOSUPRESOR CONVENCIONAL
A pesar del cumplimiento terapéutico, el 10% de los pacientes pueden empeorar1,6,11. Esta mala respuesta obliga a administrar dosis elevadas de prednisona (60 mg/día) o de prednisona (30 mg/día) y azatioprina (150 mg/día) durante 4-6 semanas, y posteriormente reducir la medicación cada mes de mejoría clinicoanalítica hasta una dosis de mantenimiento, una vez que su diagnóstico haya sido reconfirmado y excluidas otras causas de lesión hepática. En ellos pueden probarse otras alternativas terapéuticas inmunosupresoras, y estos pacientes pueden llegar a ser candidatos de trasplante hepático.
El fracaso terapéutico se observa más frecuentemente en pacientes en los que la enfermedad se inicia como HAI aguda grave o hepatitis fulminante. Hay una serie de factores implicados en una respuesta peor al tratamiento inmunosupresor convencional, como cirrosis establecida, comienzo temprano en la edad pediátrica (muchos de estos niños tiene HAI tipo 2 y algunos tienen cirrosis cuando la enfermedad se diagnostica), pacientes con HLA DR3 (genotipo DRB1* 0301), raza negra, pacientes con variantes de la HAI como síndromes de superposición, asociación con otras comorbilidades como esteatohepatitis no alcohólica o infección por el VHC, y el mal cumplimiento terapéutico.
RESPUESTA PARCIAL
El 13% de los pacientes tienen una respuesta parcial. Estos pacientes mejoran pero no llegan a entrar en remisión completa y requieren tratamiento indefinido, y además tienen el riesgo de que la enfermedad progrese. Según el consenso de la AASLD6, se debe tratar como el fracaso terapéutico. Estos pacientes pueden ser subsidiarios de alternativas terapéuticas al tratamiento convencional.
ALTERNATIVAS TERAPÉUTICAS
Aproximadamente el 10% de los pacientes con HAI no responden al tratamiento convencional con corticoides, el 13% tiene una respuesta parcial y un 13% sufre toxicidad que obliga a suspender prematuramente el tratamiento. También en una pequeña proporción de enfermos se pueden elevar de nuevo los valores de transaminasas después de una respuesta inicial beneficiosa. Las alternativas terapéuticas en estos casos son: ciclosporina, metotrexato, ciclofosfamida, micofenolato-mofetilo, tacrolimus, ácido ursodesoxicólico y budesonida. Sin embargo, hay pocos datos referentes a estas estrategias terapéuticas y se precisan más estudios que aclaren el papel de cada uno de estos fármacos en el tratamiento de la HAI.
El trasplante hepático es la última opción terapéutica en alrededor del 10% de los pacientes con HAI que no responden al tratamiento médico19,20. El trasplante hepático por esta indicación supone el 2,6 % de los trasplantes hepáticos en Europa y el 5,9% en Estados Unidos. Está indicado en enfermos con cirrosis e insuficiencia hepática avanzada, cuando surgen las complicaciones de la hipertensión portal y si la HAI se presenta como una hepatitis fulminante. El pronóstico es muy favorable, con una supervivencia a los 5 años tras el trasplante próxima al 90% y una supervivencia a los 10 años del 75%20.
La incidencia de recurrencia de la HAI postrasplante es elevada, de alrededor del 20-30%, especialmente en los pacientes que no reciben una inmunosupresión adecuada.