






La enfermedad de Marfan (EM), provocada por una mutación heterocigota en el gen que codifica la fibrilina-1 y transmitida de forma autosómica dominante, es, con una incidencia aproximada de 1caso por cada 10.000 nacidos vivos, una de las enfermedades hereditarias del tejido conectivo más frecuentes.
Desde su descripción en 1986 por Antoine-Bernard-Jean Marfan el conocimiento de la enfermedad se ha ampliado de forma progresiva, con la descripción de afectaciones oculares, esqueléticas, cardiovasculares, en el pulmón, en la piel y en los tegumentos que definen la enfermedad, recopilados en los criterios de Ghent, que en la actualidad conforman la base del diagnóstico de la enfermedad en adultos.
El diagnóstico de la EM se encuentra a veces comprometido por el hecho de que los hallazgos clínicos son dependientes de la edad, lo que condiciona dificultades en el diagnóstico en niños y en pacientes jóvenes; algunos de ellos son frecuentes en la población general no afectada; además, la EM tiene una alta penetrancia pero con una variabilidad fenotípica sustancial, y se da un solapamiento importante entre las diferentes enfermedades del colágeno, lo que dificulta a veces su distinción.
El síndrome de Marfan ha sido objeto en los últimos años y en la actualidad de un creciente interés por la detección de otras enfermedades con fenotipo marfanoide y mutaciones en el gen de la fibrilina-1, y por la aparición de un tratamiento médico y quirúrgico agresivo que ha cambiado de forma radical el pronóstico de la enfermedad.
La naturaleza multisistémica de la enfermedad requiere un manejo multidisciplinar.
Marfan's disease is provoked by a heterozygotic mutation in the gene codifying fibrillin-1 and is transmitted in an autosomic dominant form. The incidence is 1 out of every 10,000 live births, making it one of the most frequent hereditary connective tissue diseases.
Since Antoine-Bernard-Jean Marfan first described the syndrome in 1986, knowledge of this entity has progressively increased. Affected areas include the eyes, skeletal and cardiovascular systems, the lung, skin and the tissue covering the spinal cord, which have been described in the Ghent criteria, currently the basis for diagnosis.
Diagnosis of Marfan's disease can be difficult because the clinical findings are age-dependent, sometimes leading to difficulties in diagnosing children and young patients. Some of these findings are also frequent in the general population without the disease. Furthermore, the disease has high penetration but there is wide phenotypic variability and substantial overlap with the distinct collagen diseases, hampering differential diagnosis.
In recent years, interest in Marfan's syndrome has grown due to the detection of other diseases with marfanoid phenotype and mutations in the fibrillin-1 gene and the development of aggressive medical and surgical treatment that has radically changed prognosis. Because Marfan's disease is multisystemic, multidisciplinary management is required.
Las enfermedades hereditarias del tejido conectivo conforman un grupo en el que la matriz del colágeno, una de las proteínas más abundantes en el organismo, se encuentra afectada. Existen varias clasificaciones de estas enfermedades, que incluyen la enfermedad de Marfan (EM) y otras relacionadas, como el síndrome de Ehler-Danlos, la osteogénesis imperfecta, el seudoxantoma elástico y la cutis laxa.
La EM, provocada por una mutación heterocigota en el gen que codifica la fibrilina-1 (FBN1) y transmitida de forma autosómica dominante, es una de las más frecuentes, con una incidencia aproximada de 1caso por cada 10.000 nacidos vivos.
En 1896 Antoine-Bernard-Jean Marfan describió por primera vez esta entidad en una niña de 5años con varias anomalías esqueléticas1. En 1995, McKusick incluyó esta enfermedad con un patrón autosómico dominante en la categoría de enfermedades del colágeno hereditarias2. Desde entonces el conocimiento de la enfermedad se ha ampliado de forma progresiva, con la descripción de afectaciones oculares, esqueléticas, cardiovasculares, en el pulmón, en la piel y en los tegumentos que definen la enfermedad, recopiladas en los criterios de Ghent3, que en la actualidad conforman la base del diagnóstico de la enfermedad en adultos. La detección en 1991 del gen que codifica la FBN14 y que conforma la base genética de la enfermedad amplió el conocimiento global de la enfermedad y ayudó a la definición de otras enfermedades del colágeno asociadas al Marfan, algunas con mejor pronóstico que la misma enfermedad y otras con un peor pronóstico que precisa diferentes consideraciones en su seguimiento.
Puesto que la EM se asocia con una muerte prematura en pacientes sin tratamiento, la realización de un diagnóstico correcto y precoz es de gran importancia. En la actualidad el diagnóstico no se realiza de forma exclusiva con el análisis genético molecular, ya que a veces no está disponible, la detección de la mutación es imperfecta y no todas las mutaciones del gen de la FBN1 se asocian a EM.
La disección de la aorta fue la causa principal de mortalidad en estos pacientes hasta la introducción del tratamiento con betabloqueadores de forma precoz a partir de 19715 y la implantación de la cirugía profiláctica de la aorta6, así como los ensayos actuales con losartán7,8.
El síndrome de Marfan ha tenido en los últimos años y en la actualidad un aumento de interés por la detección de otras enfermedades con fenotipo marfanoide y mutaciones en el gen de la FBN1, y la aparición de un tratamiento médico y quirúrgico agresivo que ha cambiado de forma radical el pronóstico de la enfermedad6.
La naturaleza multisistémica de la enfermedad precisa de un manejo multidisciplinar.
Frecuencia y epidemiologíaLa EM (MFS, OMIM #154700) es una de las enfermedades del colágeno hereditarias más comunes, causada de forma principal por una mutación heterocigota en el gen que codifica la FBN1 (15q21.1)4, con una incidencia aproximada de 1por cada 5.000 a 1-3por cada 10.000 personas vivas, según las series9,10; afecta por igual a ambos sexos y su distribución es mundial.
EtiologíaAlteraciones molecularesLa EM es una enfermedad autosómica dominante con alta penetrancia, de forma que todos los portadores virtualmente desarrollan la enfermedad aunque con marcada heterogeneidad fenotípica11.
La mayoría de los casos de Marfan son causados por una mutación en el gen de la FBN1, y en el 75% de los casos existen antecedentes familiares. No obstante, en alrededor del 25% de los casos se detectan mutaciones de novo12. Se han descrito entre 600 y 1.000 mutaciones distintas, y la mayoría de ellas son del tipo de cambio de sentido (missense-type), alterando algún aminoácido de los 2.871 que constituyen la proteína, usualmente en los dominios homólogos a los del factor de crecimiento epidérmico (EGF), y afectando residuos de cisteína o aminoácidos implicados en el transporte de calcio13 y exclusivos de cada caso índice o familiar14,15.
La base de datos UMD FBN1(http://www.umd.be) se creó en 1995 para recopilar todas las mutaciones detectadas, y hasta el momento hay 600-1.000 mutaciones disponibles online.
Descubrir la base molecular del MFS y de las fibrilinopatías relacionadas es el objetivo de los estudios moleculares actuales, pero la extremada variabilidad clínica, la dificultad del diagnóstico clínico en algunos casos y el bajo índice de detección de mutaciones en este gen tan largo tienen un impacto negativo en los progresos.
Mutación en el gen de la FBN1El gen de la FBN1 se localiza en el cromosoma 15q21.1 y codifica una glicoproteína (profibrilina 1) que se convierte en FBN1. Está constituido primariamente por la repetición de dominios homólogos de factor de crecimiento transportador de calcio (cbEGF) y dominios que contienen 8 residuos de cisteína16. La mutación en pacientes con SMF se localiza normalmente en la unidad cbEGF14.
Aunque en general no existe una clara correlación entre el genotipo detectado y el fenotipo expresado, la correlación más significativa geno-fenotípica se encuentra en la EM neonatal, detectando la mutación en los exones 24-3217, lo que parece predecir un fenotipo más agresivo. Las mutaciones en los exones 1-10 parecen correlacionarse con formas sin dilatación aórtica, y las mutaciones en los exones 59-65 se asocian con formas cardiovasculares tardías y menores18. Las mutaciones que causan una parada prematura en los codones se asocian menos con ectopia lentis y desprendimiento de retina que las mutaciones que sustituyen un residuo de cisteína en el dominio cbEGF19. A pesar de todo esto, la variabilidad fenotípica intrafamiliar es importante en la EM, y en la mayoría de los casos no existe una correlación geno-fenotípica2,11,20, a pesar de los datos expuestos anteriormente.
Asimismo, las mutaciones en el gen de la FBN1 no son específicas de la EM y se han detectado también en otras fibrilinopatías12.
Mutaciones en el TGFbR1 y TGFbR2El receptor del TGF-beta es un heterodímero implicado en la unión entre las subunidades I y II (codificado por los genes TGFBR1 y TGFBR2); se detectan mutaciones en el síndrome de Loeys Dietz y en algunos pacientes con aneurismas familiares y disección aórtica21,22.
La FBN1 ejerce su control sobre el sendero señalado del TGF-beta: altera su activación y puede provocar una señalización y activación excesivas.
La identificación de mutaciones en el gen FBN1, el FBN2 (síndrome de Beals), el TGFBR1 y el TGFBR2 ha aumentado el conocimiento sobre la patogenia del Marfan y de otras enfermedades relacionadas, y puede ayudar en la caracterización de un mayor espectro de mutaciones asociadas a las enfermedades de colágeno genéticas12.
PatogeniaA pesar de los avances en el conocimiento de la biología molecular en la EM y en otros síndromes relacionados, el mecanismo molecular que conduce al desarrollo del fenotipo no se conoce de forma clara. El hallazgo de degeneración quística medial en la pared de la aorta de pacientes con Marfan, aunque también ocurre en otros pacientes con aneurismas torácicos23, en la EM lo hace de forma más precoz, y la identificación del gen afectado condujo a las primeras hipótesis patogénicas de la enfermedad.
La FBN1, proteína mutada en la EM, es una glicoproteína extracelular de 350 kilodaltons, muy parecida entre las diferentes especies. La polimerización extracelular de las fibrilinas como monómeros forma macroagregados llamados microfibrillas, en asociación con otras proteínas como el TGF-beta. Las microfibrillas proveen un fuerte soporte estructural y en asociación con la elastina forman las fibras elásticas, que aportan elasticidad a la estructura de forma temporal y dependiente del tejido12.
Durante mucho tiempo se pensó que la base exclusiva de la enfermedad era la pérdida de la integridad del tejido conectivo con la reducción y la fragmentación de las fibras elásticas en los tejidos afectados, lo cual ofrece una explicación de la patología aórtica, aunque no de otras manifestaciones de la enfermedad como el sobrecrecimiento del hueso largo, el engrosamiento valvular y la hipoplasia muscular24.
Otros mecanismos que se han barajado incluyen:
- •
Incremento de la señal TGF. Estudios experimentales con ratones con deficiencia de FBN1 han cambiado el modelo fisiopatológico actual de la EM. En estos modelos la deficiencia estructural de FBN1 conduce a una activación de la citocina TGF-beta, la cual tiene un papel fundamental en el desarrollo y el mantenimiento de varios tejidos. La activación de la TGF-beta parece contribuir al desarrollo de enfisema, aneurismas aórticos e hipoplasia muscular que se observa en el Marfan25,26. Pero recientemente se ha encontrado una posible correlación entre el aumento de señal de TGF-beta y el desarrollo de aneurismas. Habashi et al26 realizaron un estudio en ratones heterocigotos para la mutación del gen de la FBN1, comparando el efecto entre el losartán (bloqueador del TGF-beta) y los betabloqueadores durante 6 meses; tras el periodo de estudio en el grupo de losartán se observó de forma exclusiva una pared aórtica con arquitectura mejor conservada que en los ratones sanos, por lo que al menos en el modelo murino estos cambios podrían ser bloqueados de forma efectiva por la administración de anticuerpos anti TGF-beta26.
- •
Activación de la metaloproteinasa (MMP). Las MMP constituyen una gran familia de enzimas que procesan y degradan un gran número de sustratos, como el colágeno y la elastina, y son regulados por 4 factores tisulares inhibitorios (TIMP)27. La remodelación progresiva de los vasos arteriales en la EM se correlacionan con el aumento de la actividad de la MMP-2 y MMP-9 favoreciendo la proteólisis de la matriz extracelular. Varios estudios han demostrado la existencia de un desequilibrio entre MMP/TIMP en muestras de tejido aórtico. El tipo de MMP parece condicionar el tamaño del aneurisma; así, niveles más elevados de MMP-2 se asocian a aneurismas de tamaño más pequeño que los asociados a incremento del MMP-928.
Este concepto de la EM como un trastorno estructural del tejido conectivo que se manifiesta como anormalidades en el desarrollo que alteran la señal de las citocinas abre nuevas e inesperadas dianas terapéuticas29.
Manifestaciones clínicas y diagnósticoEl diagnóstico clínico y molecular en la EM se ve afectado por varios factores:
- •
Los hallazgos clínicos son dependientes de la edad, lo que condiciona dificultades en el diagnóstico en niños y pacientes jóvenes.
- •
Algunos de los hallazgos son frecuentes en la población general no afectada, como el prolapso de la válvula mitral.
- •
La EM tiene una alta penetrancia, pero con una variabilidad fenotípica sustancial.
- •
Hay, además, un solapamiento importante entre las diferentes enfermedades del colágeno, lo que dificulta a veces su distinción.
- •
Por último, en la actualidad no se realiza el diagnóstico con el análisis genético molecular de forma exclusiva, ya que a veces no está disponible, la detección de la mutación es imperfecta y no todas las mutaciones del gen de la FBN1 se asocian a EM, como se ha comentado en el apartado de genética.
En 1986 se propusieron los primeros criterios diagnósticos de la enfermedad30. Tras el descubrimiento de la base molecular de la enfermedad4 en 1995 se propusieron los actuales criterios diagnósticos de la enfermedad, los criterios de Ghent5 (tabla 1), de manera que el diagnóstico clínico en adultos debe realizarse basándose en estos, aunque no son útiles en niños y en adultos jóvenes. El diagnóstico de EM con estos criterios facilita el reconocimiento de este síndrome aneurismático genético y mejora su tratamiento. Tales criterios han demostrado ser eficaces incluso tras la mejora de las técnicas de biología molecular, consiguiendo la confirmación diagnóstica hasta en el 90% de los casos31.
Criterios de Ghent clásicos
| Sistema afecto | Requisito clasificatorio de sistema afectado cumpliendo criterio mayor de afectación | Requisito clasificatorio para considerar el sistema afectado |
| Sistema esquelético | Al menos 4 de los siguientes:1. Pectum carinatum2. Pectum excavatum quirúrgico3. Asimetría segmentos corporales y brazos (brazada >1,05)4. Signos de Wrist y Thumb+5. Escoliosis >20°6. Extensión reducida codos (<170°)7. Desplazamiento maléolo medial8. Profusión acetabular | Al menos 2 criterios mayores o uno mayor y dos menores de los siguientes:1. Pectum excavatum moderado2. Hipermovilidad articular3. Paladar arqueado/dientes apiñados4. Características faciales típicas (dolicocefalia, hipoplasia malar, enoftalmos, retrognatia, fisura palpebral inclinada hacia abajo) |
| Sistema ocular | Luxación del cristalino | Al menos dos de las siguientes:1. Anormalidades de los compartimentos corneales2. Aumento longitud axial del globo ocular3. Iris hipoplásico o hipoplasia de los músculos ciliares con disminución de la miosis |
| Sistema cardiovascular | Al menos uno de los siguientes:1. Dilatación de la aorta ascendente con afectación de los senos de Valsalva2. Disección de la aorta ascendente | Al menos uno de los siguientes criterios menores:1. Prolapso de válvula mitral con o sin regurgitación.2. Dilatación de las arterias pulmonares <40 años sin causa valvular conocida.3. Calcificación anillo mitral <40 años.4. Dilatación o disección de la aorta descendente o abdominal<50 años. |
| Sistema pulmonar | Ninguno | Al menos uno de los criterios menores:1. Neumotorax espontáneo2. Bullas apicales |
| Piel y tegumentos | Ninguno | Al menos uno de los criterios menores:1. Estrías marcadas sin causa aparente2. Hernias recurrentes o eventraciones |
| Duramadre | Ectasia dural | |
Para establecer el diagnóstico de EM se precisa de al menos un criterio mayor en al menos dos sistemas afectados y un tercer sistema afectado. En caso de historia familiar o genética compatible (definida por la presencia de una mutación en el FBN1 conocida asociada en EM o herencia de un haplotipo FBN1 conocida o inequívoca asociación al diagnóstico de Marfan en una familia) es suficiente con un criterio mayor de un órgano o sistema y/o la afectación de un segundo (tabla 1). Los criterios no pueden aplicarse antes de los 18 años ya que la enfermedad habitualmente se expresa de forma incompleta32.
Manifestaciones clínicas de la enfermedadLa mayoría de las manifestaciones clínicas, ordenadas por órganos o sistemas afectados, conforman los criterios de Ghent clásicos (tabla 1):
Sistema esqueléticoConforma el llamado hábito marfanoide, caracterizado por aracnodactilia, aumento del segmento corporal inferior sobre el superior y facies típica, caracterizada por dolicocefalia, hipoplasia malar, enoftalmos, retrognatia y fisuras palpebrales con inclinación inferior. Para considerar el órgano afectado en los criterios de Ghent se precisa de dos criterios mayores o uno mayor y dos menores, que se describen en la tabla 1.
Sistema ocularLos ojos son órganos que se afectan de forma típica, y la luxación de cristalino es un criterio mayor diagnóstico. La luxación de cristalino afecta hasta al 50% de los pacientes, suele ser superior y temporal y aparece en cualquier momento de la evolución de la enfermedad. Los criterios menores (se requieren al menos dos para definir el órgano afectado) incluyen: anormalidades en los compartimentos corneales (queratometría), aumento axial de la longitud del globo ocular (medida por ecografía), iris hipoplásico o musculatura ciliar hipoplásica con disminución de la miosis.
Otras afectaciones que se observan son: miopía (por elongación del globo ocular y ambliopía), cataratas (nucleares escleróticas) en <50 años, glaucoma en <50 años y desprendimiento de retina.
Para la exploración del aparato ocular en el síndrome de Marfan se precisa el examen con lámpara de hendidura, con dilatación pupilar, queratometría y biometría ocular para medición del diámetro anteroposterior del ojo11,33.
Sistema cardiovascularLa afectación de este sistema es la causa principal de morbimortalidad en la EM. Los criterios mayores incluyen:
- •
Dilatación de la raíz aórtica, con afectación de los senos de Valsalva, con una prevalencia del 70-80%. Se manifiesta a edad temprana y tiende a ser mayor en hombres.
- •
Disección aórtica, con afectación de la aorta ascendente.
Otras alteraciones del sistema cardiovascular que conforman los criterios menores de Ghent incluyen: prolapso de válvula mitral (prevalencia del 55-69% en EM frente al 2% de la población general)10, dilatación de la arteria proximal pulmonar principal en ausencia de estenosis pulmonar periférica u otra causa que lo explique, calcificación del anillo mitral (pacientes <40 años) y dilatación de la aorta abdominal o descendente torácica (<50 años), que afecta al 10% de los pacientes con EM y es en la actualidad la causa más importante de mortalidad en ellos, tras los avances en el tratamiento médico y la cirugía profiláctica de la aorta ascendente11,34.
La ecocardiografía transtorácica (ETT) es la prueba fundamental para la evaluación y el seguimiento de la afectación cardiovascular. Para la evaluación de la dilatación aórtica, la proyección de elección es el eje largo de la ventana paraesternal35; todas las medidas deben ser estrictamente perpendiculares al eje de la aorta y comparadas con nomogramas dependiendo de la edad y de la superficie corporal del paciente35. La dilatación aórtica se define como un diámetro normalizado mayor de la media más dos desviaciones estándar (Z score>2). Pero a veces los nomogramas no son válidos en pacientes con tallas superiores al percentil 95, pudiendo sobreestimar en estos pacientes el diámetro de la aorta. En estos casos, algunos autores proponen la medición del ratio entre el diámetro de los senos de Valsalva y del anillo aórtico11,36,37; por lo general este ratio permanece constante en personas sanas y es independiente de la edad y de la superficie corporal, pudiendo ser de ayuda en la evaluación en niños (un ratio >1,45 predeciría la existencia de dilatación aórtica con una sensibilidad del 82% y una especificidad del 100%)37. No obstante este ratio, aun siendo de ayuda, no está suficientemente validado y tiene falsos positivos, como pacientes con anillo aórtico estrecho.
En pacientes con mala ventana ecocardiográfica tanto la tomografía computarizada (TC) como la resonancia magnética (RM) son de ayuda para el diagnóstico y para el seguimiento en pacientes con cirugía aórtica, así como para evaluar el resto de la aorta (que puede estar afectada hasta en el 10% de los pacientes).
Ectasia duralEn los criterios clásicos de Ghent la ectasia dural conforma un criterio mayor para el diagnóstico de la EM. Se trata de un ensanchamiento del saco dural y del canal espinal, a veces con afectación de la raíces nerviosas, con una prevalencia en Marfan del 65-92%. También se ha descrito asociada a Ehler-Danlos, neurofibromatosis tipo I, espondilitis, traumatismo, escoliosis o tumores2,11. Precisa de TC o RM para el diagnóstico cualitativo y cuantitativo. La TC es menos precisa, y para el diagnóstico se requiere la presencia o no de dilatación del saco dural, erosión central del cuerpo vertebral en el plano sagital o meningocele. La evaluación cuantitativa es más sensible pero compleja, y requiere el ratio entre la medida del diámetro del saco dural y el diámetro anteroposterior de cada cuerpo vertebral desde L5 a S1 (conocido como ratio del saco dural). Un ratio del saco dural en L3 >0,47 o en S1 >0,57 diagnostica la ectasia dural en adultos, y ha sido validado en niños con Marfan con una sensibilidad del 95% y una especificidad del 98%38,39. Los síntomas más comunes son: lumbalgia, cefalea, debilidad y disminución de la sensibilidad, y ocasionalmente dolor rectal y púbico. Se agrava en posición supina y mejora al acostarse; su gravedad aumenta con la edad, y es severa solo en menos del 20% de los pacientes11,38.
Criterios de Ghent modificadosA pesar de haber demostrado su validez a lo largo de su aplicación, los criterios de Ghent presentan algunos problemas. En primer lugar, los criterios diagnósticos actuales no han sido suficientemente validados; en segundo lugar, no pueden ser aplicados en niños, y en tercer lugar, requieren a veces de estudios caros y especializados. El descubrimiento de una expresión clínica variable y de las enfermedades relacionadas con el Marfan que han sido incluidas recientemente en el diagnóstico diferencial afecta a los métodos diagnósticos actuales, dificultando el diagnóstico. Además el diagnóstico de la EM, tanto si se ha establecido de forma correcta como si no, puede ser estigmatizador, alterar las aspiraciones futuras, restringir las oportunidades de los seguros de salud y causar alarma psicosocial. Todos estos hechos llevan a la revisión de los criterios de Ghent por un panel internacional de expertos, que confiere un mayor peso específico a las manifestaciones cardiovasculares y a la subluxación del cristalino como hallazgos clínicos cardinales40. En los criterios modificados de Ghent (tabla 2), en ausencia de historia familiar, la presencia de estas dos manifestaciones es suficiente para el diagnóstico. En ausencia de alguna de estas dos, o de ambas, la presencia de la mutación FBN1 clásica y la combinación de una serie de hallazgos sistémicos (tabla 2) son necesarias para diagnóstico. En estos criterios modificados se considera, además, el diagnóstico en niños, definiendo el concepto de enfermedad del tejido conectivo inespecífica y síndrome de Marfan emergente o potencial (tabla 3) y diagnósticos alternativos en adultos como el fenotipo MASS o la luxación congénita de cristalino en pacientes con mutaciones en el gen FBN1 u otras colagenopatías con hallazgos similares no asociadas a la mutación en el gen FBN1, como el síndrome de Ehlers-Danlos tipo IV, el síndrome de Loeys- Dietz y el síndrome de Shprintzen-Goldberg.
Criterios de Ghent modificados
| Ausencia de historia familiar | Presencia de historia familiar |
| Ao (Z≥2) y LC=EM* | LC e historia familiar EM=EM |
| Ao (Z≥2) y bonafide FBN1=EM | Puntuación sistémica (≥7) e historia familiar EM=EM* |
| Ao (Z ≥ 2) y puntuación sistémica (≥7)=EM* | Ao (Z≥2) en >20 años o Ao (Z≥3) en <20 años e historia familiar EM=EM* |
| LC y FBN1 con conocida asociación a Ao=EM | |
| LC con o sin afectación sistémica y con FBN1 no asociada a afectación aórtica o ausencia FBN1=luxación del cristalino familiar | |
| Ao (Z<2) y puntuación sistémica (≥5 con al menos un hallazgo esquelético) sin LC=fenotipo MASS | |
| PVM y Ao (Z<2) y puntuación sistémica (<5) sin LC=síndrome de prolapso de la válvula mitral | |
| Hallazgos sistémicos | Puntuación | |
| Signo de Wrist y Thumb | 3 | Signo de Wrist o thumb –1 |
| Pectum carinatum | 2 | Pectum excavatum o asimetría torácica –1 |
| Deformidad en el retropié | 2 | Pies planos –1 |
| Neumotórax | 2 | |
| Ectasia dural | 2 | |
| Protrusión acetabular | 2 | |
| Reducción del segmento superior/inferior y aumento de la brazada sin escoliosis severa | 1 | |
| Escoliosis y/o cifosis toracolumbar | 1 | |
| Hallazgos faciales (3/5) | 1 | Dolicocefalia, enoftalmos, fisura parpebral con inclinación inferior, hipoplasia malar, retrognatia |
| Estrías en la piel | 1 | |
| Miopía >3 dioptrías | 1 | |
| Prolapso válvula mitral | 1 |
EM*: diagnóstico en ausencia de datos de SLD, ED tipo IV, y SGS, realizando si procede TGFBR1 y 2, COl3a o estudio del colágeno; Ao: diámetro aórtico desde los senos de Valsalva medido con Z score o disección de la aorta; LC: luxación de cristalino; EM: enfermedad de Marfan; PVM: prolapso de la válvula mitral; SLD: síndrome de Loeys-Dietz; SGS: síndrome de Shprintzen-Goldberg; ED: síndrome de Ehlers-Danlos.
Máximo total: 20 puntos; score ≥ 7 indica afectación sistémica.
Definición de EM emergente y enfermedad del tejido conectivo inespecífica
| Síndrome de Marfan emergente | Pacientes jóvenes con historia familiar y estudio genético no diagnóstico y score sistémico <7 o Ao Z>3 |
| Enfermedad del tejido conectivo inespecífica | Pacientes jóvenes sin historia familiar, score sistémico <7 o Ao Z<3, con solo un sistema afectado (p. ej., esquelético) |
Ao: diámetro aórtico desde los senos de Valsalva medido con Z score.
Los autores de los criterios modificados creen que las nuevas guías retrasarán el diagnóstico definitivo de la EM pero disminuirán el riesgo de falsos diagnósticos prematuros.
Estudio genético en el diagnóstico de la enfermedad de MarfanEn la actualidad el diagnóstico de la EM se realiza por criterios clínicos (criterios de Ghent), que en la mayoría de los casos son suficientes para establecer el diagnóstico. En algunos casos no es fácil el diagnóstico con los criterios clínicos.
Por esto, en pacientes que cumplen los criterios de Ghent se puede hacer un diagnóstico certero, pero en los que no lo hacen, si presentan manifestaciones sistémicas compatibles precisan evaluación clínica completa, así como seguimiento oftalmológico y ecocardiográfico en la mayoría de los casos.
En estas situaciones de diagnóstico clínico dudoso, el análisis genético del FBN1 podría ayudar al diagnóstico. En la práctica, no obstante, la compleja estructura del gen FBN1 y la diferente metodología para su detección varían sensiblemente la sensibilidad y la especificidad de la muestra en las diferentes series12. En la actualidad parece ser que la detección de la mutación por cromatografía líquida de alto rendimiento (DHPL) es la más eficiente a pesar de sus falsos positivos12. El factor más importante que influye en la detección de la mutación parece ser la homo o heterogeneidad clínica que el paciente presente, como demuestran diferentes estudios en los que la incidencia de la mutación FBN1 es significativamente superior en los pacientes que cumplen los criterios de Ghent: hasta en el 66%, frente al 5% en enfermedades relacionadas con el Marfan41. También puede ser de ayuda la identificación del gen FBN1 en un niño que no cumple los criterios mayores oculares o cardiovasculares, independientemente de la historia familiar, identificando a los que deben continuar controles o no (si se detecta el gen en la familia o existen antecedentes familiares). La identificación del gen FBN1 en un paciente que no cumple criterios de Marfan pero que puede tener una enfermedad relacionada es importante para el seguimiento, ya que en estos casos se aconseja seguimiento cardiovascular por el riesgo per se que presentan de dilatación aórtica (tabla 4).
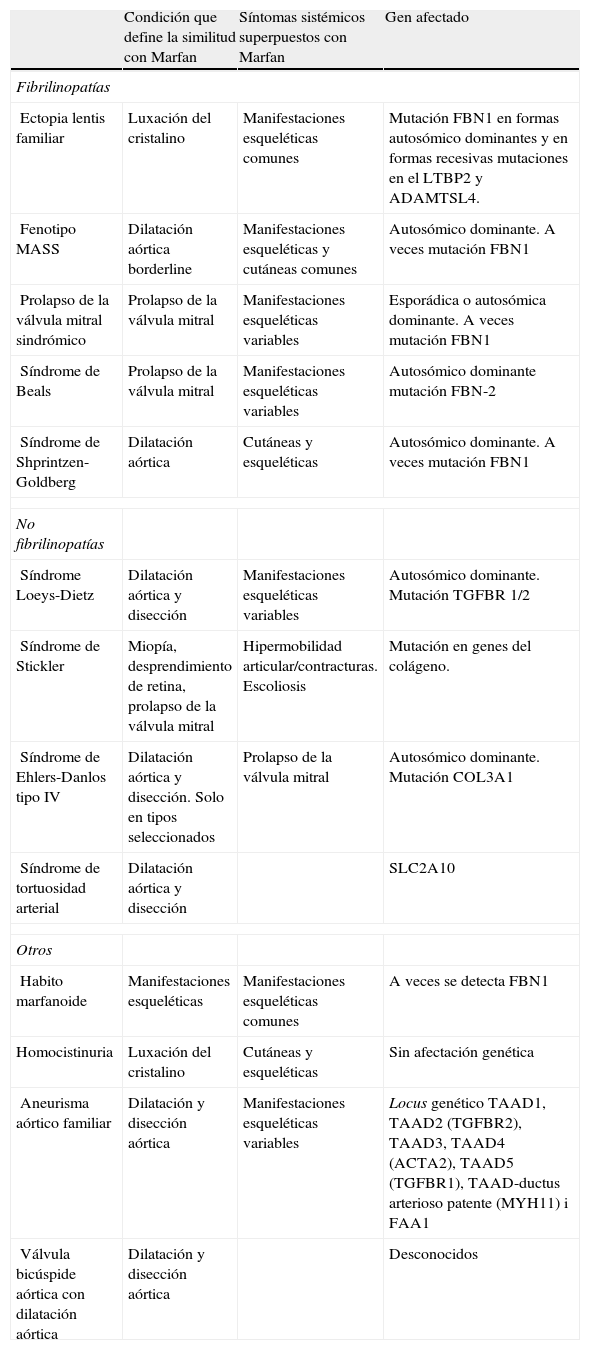
Diagnóstico diferencial con la enfermedad de Marfan
| Condición que define la similitud con Marfan | Síntomas sistémicos superpuestos con Marfan | Gen afectado | |
| Fibrilinopatías | |||
| Ectopia lentis familiar | Luxación del cristalino | Manifestaciones esqueléticas comunes | Mutación FBN1 en formas autosómico dominantes y en formas recesivas mutaciones en el LTBP2 y ADAMTSL4. |
| Fenotipo MASS | Dilatación aórtica borderline | Manifestaciones esqueléticas y cutáneas comunes | Autosómico dominante. A veces mutación FBN1 |
| Prolapso de la válvula mitral sindrómico | Prolapso de la válvula mitral | Manifestaciones esqueléticas variables | Esporádica o autosómica dominante. A veces mutación FBN1 |
| Síndrome de Beals | Prolapso de la válvula mitral | Manifestaciones esqueléticas variables | Autosómico dominante mutación FBN-2 |
| Síndrome de Shprintzen-Goldberg | Dilatación aórtica | Cutáneas y esqueléticas | Autosómico dominante. A veces mutación FBN1 |
| No fibrilinopatías | |||
| Síndrome Loeys-Dietz | Dilatación aórtica y disección | Manifestaciones esqueléticas variables | Autosómico dominante. Mutación TGFBR 1/2 |
| Síndrome de Stickler | Miopía, desprendimiento de retina, prolapso de la válvula mitral | Hipermobilidad articular/contracturas. Escoliosis | Mutación en genes del colágeno. |
| Síndrome de Ehlers-Danlos tipo IV | Dilatación aórtica y disección. Solo en tipos seleccionados | Prolapso de la válvula mitral | Autosómico dominante. Mutación COL3A1 |
| Síndrome de tortuosidad arterial | Dilatación aórtica y disección | SLC2A10 | |
| Otros | |||
| Habito marfanoide | Manifestaciones esqueléticas | Manifestaciones esqueléticas comunes | A veces se detecta FBN1 |
| Homocistinuria | Luxación del cristalino | Cutáneas y esqueléticas | Sin afectación genética |
| Aneurisma aórtico familiar | Dilatación y disección aórtica | Manifestaciones esqueléticas variables | Locus genético TAAD1, TAAD2 (TGFBR2), TAAD3, TAAD4 (ACTA2), TAAD5 (TGFBR1), TAAD-ductus arterioso patente (MYH11) i FAA1 |
| Válvula bicúspide aórtica con dilatación aórtica | Dilatación y disección aórtica | Desconocidos | |
El diagnóstico diferencial de la EM es amplio (tabla 4) y fundamentalmente incluye otras enfermedades del colágeno con o sin afectación del gen FBN1 y otras enfermedades que tienen hallazgos comunes con los diferentes órganos que se afectan en la EM29. Por el motivo de consulta con que el enfermo es enviado a la consulta para descartar EM, podemos diferenciarlas en enfermedades con afectación cardiovascular, oftalmológica y sistémica similar.
Afectación cardiovascularEl diagnóstico diferencial incluye:
- •
Fenotipo MASS. Se refiere al acrónimo prolapso de válvula mitral; dilatación de la raíz aórtica, la cual suele estar en el límite superior de la normalidad pero con escasa progresión a aneurisma o predisposición a la disección29,40; afectación cutánea (skin) con aparición de estrias no relacionadas con la ganancia de peso; y afectación esquelética (skeletal), con manifestaciones comunes con EM (incluyendo escoliosis, malformaciones torácicas e hipermobilidad articular). El fenotipo MASS presenta herencia dominante, detectándose a veces mutaciones en el FBN1. En los nuevos criterios de Ghent40 se define como diámetro aórtico desde los senos de Valsalva medido con Z score (Ao z) <2, al menos un hallazgo esquelético y puntuación sistémica ≥5. Si el gen FBN1 es positivo, podría ser un síndrome de Marfan emergente.
- •
Síndrome de prolapso de la válvula mitral. Afecta al 1% de la población, con limitados hallazgos sistémicos (puntuación <5), incluido pectum excavatum, escoliosis y aracnodactilia. La dilatación aórtica y la luxación del cristalino excluyen el diagnóstico29,40.
- •
Válvula bicúspide aórtica. La prevalencia es del 1% en la población general. Un subconjunto de individuos puede presentar aneurisma aórtico ascendente, y en estas familias se observa afectación esquelética, como escoliosis29,40.
- •
Síndrome de Loeys-Dietz. Descrito en 200521, es una enfermedad del colágeno hereditaria autosómica dominante causada por afectación del TGFBR1 y del TGFBR2. Más agresiva que el síndrome de Marfan y con mayores complicaciones en el embarazo, se caracteriza por un alto riesgo de disección y rotura aórtica a edades tempranas, sin parámetros previos que prevean la situación. Se han descrito dos subtipos; el tipo 1 es más agresivo y se caracteriza por afectación craneofacial, mientras que el tipo 2 se caracteriza por úvula bífida. Aunque los individuos afectados no son inapropiadamente altos ni tienen extremidades desproporcionadamente largas, presentan hallazgos comunes con el Marfan, como aracnodactilia, dilatación aórtica y aneurismas, prolapso de válvula mitral, pectum excavatum o carinatum, escoliosis, cifosis, articulaciones flexibles, pies planos y ectasia dural. De forma exclusiva presentan la tríada: tortuosidad arterial, aneurismas y disecciones en arterias diferentes de la aorta; hipertelorismo, y úvula ancha o bífida/paladar hendido. Otras manifestaciones incluyen: defectos del corazón al nacimiento; afectación cutánea con hematomas espontáneos, amplias cicatrices, textura suave de la piel, y piel translúcida; problemas gastrointestinales que incluyen diarrea, dolor abdominal, hemorragia intestinal o perforación; rotura del útero durante el embarazo; inestabilidad o malformaciones de la columna y el cuello y osteoporosis21,42,43.
- •
Ehlers-Danlos tipo IV. Enfermedad autosómica dominante producida por una mutación en el gen COL3A44. No tiene especial predisposición a la afectación del arco aórtico, aunque puede afectar a la aorta en todo su recorrido y a las arterias de mediano calibre45. Se caracteriza por dismorfia facial con o sin acrogeria, síntomas cutáneos (que incluyen equimosis y hematomas) y fundamentalmente por sus complicaciones, que afectan al 25% de los pacientes antes de los 20 años y al 80% a la edad de 40, e incluyen: complicaciones vasculares, con roturas arteriales y disección con dificultad para la reparación quirúrgica, fístulas carótido-cavernosas, accidentes isquémicos en jóvenes y hemorragias intracraneales; complicaciones digestivas, con perforación espontánea del colon sigmoide, rotura del bazo y del hígado y riesgo de dehiscencia de suturas, y complicaciones obstétricas, con rotura uterina o vascular durante el embarazo (mortalidad materna del 12%)44. Otros tipos de Ehler Danlos que también pueden presentar afectación cardiovascular más o menos grave son el Ehlers-Danlos tipo cifoescoliosis y el subtipo valvular.
- •
Síndrome de tortuosidad arterial. Se caracteriza por tortuosidad grave, estenosis y aneurismas de la aorta y arterias de mediano calibre29.
- •
Síndrome de aneurisma torácico y disección familiar. Hace referencia a la predisposición genética, a veces con herencia autosómica dominante (aunque con baja penetrancia y expresión variable), a aneurismas de la aorta torácica o disecciones en ausencia de otros síntomas de Marfan o Loeys-Dietz. La edad de presentación es más temprana que en los aneurismas esporádicos (56,8 vs 64,3 años). Se asocia a los siguientes loci22,46: TAAD1, TAAD2 (TGFBR2), TAAD3, TAAD4 (ACTA2), TAAD5 (TGFBR1), TAAD-ductus aterioso persistente (MYH11) y FAA1. Como conclusión, se debe realizar estudio a los familiares de primer grado en cualquier paciente con disección aórtica (padres, hermanos e hijos) de aneurismas familiares o cualquier otra condición que cause disección aórtica12,46.
Afectación ocular con otras enfermedades, que incluyen luxación del cristalino:
- •
Ectopia lentis familiar. Síndrome genéticamente heterogéneo, autosómico dominante, causado por mutaciones en el FBN147 y formas recesivas causadas por mutaciones en el LTBP248 y ADAMTSL449. Asocia afectación ocular y esquelética, diferenciándose de la EM emergente por el seguimiento clínico, incluido ETT frecuentes, sobre todo en pacientes con mutación del FBN1.
- •
Ectopia lentis et pupillae. Enfermedad autosómica recesiva en la que están presentes remanentes de la membrana pupilar. No presenta afectación cardiovascular o esquelética.
- •
Síndrome de Weill-Marchesani. Los afectados presentan estatura baja, braquidactilia y rigidez articular, además de anormalidades oculares. Es una enfermedad transmitida con herencia autosómica dominante con afectación del gen FBN150 y recesiva, con afectación del ADAMTS1051.
- •
Homocistinuria. Metabolopatía que incluye retraso mental y trombosis. Se puede excluir por análisis de aminoácidos en orina. La luxación del cristalino en la homocistinuria suele ser inferior.
- •
Síndrome de Stickler, llamado también artro-oftalmopatía hereditaria progresiva. Asocia, además de hábito marfanoide, afectación ocular con subluxación poco habitual, pero con degeneración vítrea, glaucoma, cataratas y desprendimiento de retina52. Es una enfermedad genéticamente heterogénea, con al menos 5 subgrupos con mayor o menor riesgo de complicación oftalmológica, producidos por una mutación en el gen que codifica el colágeno tipo II (COL2A1) en el tipo 1; en los tipos 2 y 3 se produce una afectación del colágeno tipo XI.
- •
Síndrome de Beals o aracnodactilia contractural congénita. Fibrilinopatía autosómica dominante asociada a mutación en el FBN2 caracterizada por hábito marfanoide y aracnodactilia, orejas arrugadas y contracturas en las articulaciones mayores; la talla suele ser baja. Aunque se ha descrito dilatación de la aorta en los senos de Valsalva, esta no predispone a dilatación progresiva de la misma y disección53.
- •
Síndrome de Shprintzen-Goldberg. Presenta rasgos sistémicos comunes al síndrome de Marfan, como alteraciones faciales, pectum excavatum o carinatum, escoliosis y aracnodactilia, así como una alta incidencia de retraso cognitivo con baja frecuencia de enfermedad vascular54.
- •
Síndrome de Lujan- Fryns, o síndrome de hábito marfanoide y retraso mental asociado al cromosoma X. Incluye hábito marfanoide, retraso mental, dismorfia facial y alteraciones en el comportamiento55.
- •
CATSHL. Acrónimo de campilodactilia, estatura alta y pérdida de audición (campilodactilia, tall stature, hearing loss).
La expectativa de vida en la EM, condicionada fundamentalmente por el pronóstico cardiovascular, ha pasado de los 45 años en 197256 a los 72 años en 19956, en parte gracias al planteamiento de la cirugía profiláctica de la aorta, y en concreto a la implantación de la técnica de Bono Bentall.
Tratamiento farmacológico- •
Betabloqueadores. Desde 19715, año en que se publicó el primer ensayo con betabloqueadores en pacientes con Marfan, se han realizado varios estudios en adultos sobre la eficacia de los betabloqueadores en pacientes con Marfan56–58 basándose en las características básicas de estos fármacos, que disminuyen la presión del pulso y la contractibilidad miocárdica y mejoran las propiedades elásticas de la aorta, sobre todo con diámetros <40mm59,60. En la actualidad, a pesar de las escasas evidencias actuales y el pequeño o escaso impacto en la supervivencia, el tratamiento profiláctico con betabloqueadores está indicado de forma precoz tanto antes como después de la cirugía aórtica, y también está indicado en niños por extrapolación de datos con adultos, precisando en la actualidad un ensayo aleatorizado para mantener o no la indicación.
Desde hace 5 años existen evidencias de que el tratamiento con antagonistas de los receptores de la angiotensina II (ARA II) pudiera ser una alternativa a los betabloqueadores, pudiendo tener un papel adicional protector sobre la pared aórtica. Habashi et al26 encontraron, en ratones con mutación en el gen de la FBN1, que el losartán podía prevenir la dilatación de la aorta. En la actualidad están en marcha varios ensayos clínicos para demostrar su eficacia7,8.
También se han realizado estudios aislados con otros antihipertensivos, como los antagonistas del calcio y los inhibidores de la enzima de conversión de angiotensina (IECA)60,61.
En individuos con aneurisma abdominal la doxicilina ha presentado resultados prometedores, aunque estos datos no han sido extrapolados o estudiados en pacientes con Marfan62.
Cirugía profiláctica de la aorta ascendenteEn pacientes con disección de la aorta tipo A se realiza cirugía urgente. El riesgo de disección aórtica correlaciona con el diámetro de la raíz de la aorta y de la aorta ascendente, por lo que la cirugía profiláctica en adultos y en niños (en la población pediátrica el riesgo no está claramente establecido, extrapolándose los datos de la población adulta) debe considerarse cuando el diámetro de la raíz de la aorta desde los senos de Valsalva es mayor de 5cm (diámetro de la aorta/área de superficie corporal mayor o igual que 4,25cm/m2)15. Otras indicaciones o consideraciones para la realización de la cirugía profiláctica incluyen: historia familiar de disección precoz, velocidad de crecimiento aórtico >5mm/año, severidad de la regurgitación aórtica, asociación de afectación mitral, disfunción ventricular, planificación de embarazo o deseo de operación sin válvula.
En la cirugía profiláctica, inicialmente, se reseca el segmento dilatado de la aorta ascendente y se examina la válvula aórtica. Si la válvula aórtica está afectada, se utiliza la técnica de Bono- Bentall, en la que se reseca la válvula aórtica y se utiliza un injerto de dacrón con una válvula mecánica o biológica, que se sutura al anillo aórtico. Esta técnica es la de elección si la raíz aórtica tiene un diámetro superior a 60mm, si existe insuficiencia aórtica moderada-severa, en prolapsos asimétricos de varias cúspides y si hay múltiples fenestraciones de los velos62. Si no hay afectación significativa de la válvula aórtica, se utiliza alguna de las técnicas que permiten preservar la válvula y evitar la anticoagulación indefinida; todas las técnicas son igualmente efectivas, aunque con diversos índices de reoperación posterior62,63. En la técnica de remodelación (técnica de Yacoub o David II) los senos afectados se resecan, se crean tres neosenos en el injerto de dacrón y, posteriormente, este se sutura a la pared aórtica residual y al anillo aórtico. Esta técnica permite la preservación de la función del anillo aórtico, y además se forman los neosenos. En la técnica de reimplantación (técnica de David I) los senos afectados se resecan y posteriormente la válvula intacta, las comisuras y el anillo aórtico se suturan dentro del injerto de dacrón. Esta técnica previene la dilatación posterior del anillo al fijarlo al injerto de dacrón, por lo que es una técnica con mejores resultados a largo plazo y, por ello, la de elección en este tipo de pacientes62. Después de que el injerto se haya suturado a la raíz aórtica, el otro extremo del injerto se sutura a la aorta y las arterias coronarias se reimplantan en el injerto de dacrón en la posición adecuada.
Con la técnica de Bono- Bentall se consigue una supervivencia del 93%, 91%, 84%, 75% y 59% a los 1, 2, 5, 10 y 20 años, respectivamente, tras la cirugía, con una mortalidad perioperatoria del 1,5% en los centros con experiencia62,64,65, y en la actualidad la causa principal de muerte es la rotura o disección de otro segmento de la aorta66. El riesgo de reoperación fue del 2,5%, el 12,9% y el 32,9% a los 1,5 y 10 años de la cirugía, respectivamente; el aumento de la presión sistólica fue predictor de muerte tras la cirugía, y el índice de masa corporal y el tabaquismo fueron predictores de reoperación65, lo que indica la importancia del seguimiento y control de los factores de riesgo cardiovascular en los pacientes con síndrome de Marfan.
Todos los pacientes sometidos a cirugía de la aorta ascendente deben ser monitorizados de forma estricta y continuar tratamiento con betabloqueadores, salvo contraindicación. El arco aórtico, el ostium coronario y la aorta descendente pueden afectarse posteriormente, con dilatación progresiva, y está indicada la cirugía electiva de la aorta descendente si aparecen síntomas, si el crecimiento es mayor de 5-10mm/año o si el diámetro es >55mm; el riesgo de paraplejia en centros especializados es inferior al 1-2%67.
Cambios en el estilo de vidaLas recomendaciones han sido realizadas por un panel de expertos, basándose fundamentalmente en la restricción del ejercicio extenuante68. Como principio general, pueden participar en ejercicios recreativos (no competitivos) de baja a moderada intensidad (de 4 a 6 equivalentes metabólicos). Así, actividades de bajo a moderado riesgo incluirían andar, bicicleta estática y dobles de tenis; actividades de riesgo intermedio, footing, fútbol y motociclismo, y actividades de alto riesgo (que deben ser evitadas) incluyen culturismo, escalada y surf.
Guías de manejo de EM en el embarazoEl embarazo y el puerpuerio son periodos de alto riesgo para la disección y la rotura aórtica en mujeres con Marfan, debido al estrés en la pared arterial secundario a la circulación hiperdinámica e hipervolémica y al efecto estrogénico, que pueden aparecer independientemente del diámetro de la raíz aórtica, e incluso con diámetros normales69. No obstante, el riesgo de rotura aumenta con el diámetro de la aorta, y es del 1% en mujeres con diámetros <40mm y del 10% en mujeres con diámetros >40mm69,70.
Debe tenerse en cuenta, por tanto, el riesgo materno de complicación cardiovascular y el de complicaciones obstétricas, que incluyen parto prematuro en el 15% (por rotura prematura de membranas e incompetencia cervical) y mortalidad fetal y neonatal combinada del 7%. Por otra parte, debe realizarse consejo genético previo al embarazo, consejo individualizado y estricto control del embarazo70. En el embarazo se recomienda labetalol o metoprolol, ya que el atenolol parece disminuir el crecimiento fetal.
Consejo genéticoEl síndrome de Marfan es una enfermedad con herencia autosómica dominante, y las parejas con un cónyuge afectado tienen una posibilidad de transmisión del 50%. Las parejas con deseo de descendencia deben someterse a consejo genético, y puede realizarse reproducción asistida con diagnóstico genético preimplantacional, previo a la transferencia fetal uterina. También se puede realizar diagnóstico prenatal, con biopsia coriónica a las 10-12 semanas de gestación o amniocentesis en la semana 16, para detectar afectación fetal, siempre que se haya detectado la mutación que produce la afectación familiar71.
Guías de manejo y seguimientoExisten excelentes guías de manejo y seguimiento tanto para pacientes con EM como para pacientes con síndrome de Marfan emergente y enfermedades relacionadas40,62. En todas ellas recomiendan:
- •
Ecocardiogramas anuales y semestrales en pacientes con diámetros aórticos mayores de 4,5cm o con rápida velocidad de crecimiento. En jóvenes menores de 20años también deben realizarse de forma anual, y en adultos con diámetros repetidamente normales se puede realizar cada 2-3años.
- •
Tratamiento farmacológico. Iniciar tratamiento con betabloqueadores de forma precoz. Otros tratamientos, incluidos los ARA II, precisan de más estudios para sustituir a los betabloqueadores.
- •
En pacientes con disección aórtica aguda tipo A, cirugía urgente y profiláctica en los que presentan una dilatación de la aorta >5cm desde los senos de Valsalva o >4cm en mujeres previo al embarazo.
- •
En pacientes con disección de la aorta tipo B (aorta descendente) se aconseja cirugía si existe dolor intratable, isquemia de órganos o miembros inferiores, diámetro de la aorta >5,5cm o rápido aumento. A ser posible la cirugía debe ser abierta, evitando —salvo alto riesgo quirúrgico— las prótesis endovasculares por falta de experiencia67. Se aconsejan pruebas de imagen de forma regular con TC o RM en adultos y en pacientes con cirugía de la raíz aórtica.
- •
Reparación quirúrgica o recambio de la válvula mitral si existe regurgitación mitral severa sintomática, dilatación progresiva o disfunción ventricular.
- •
Profilaxis de la endocarditis. En las guías actuales de profilaxis de la endocarditis72 no se incluye la regurgitación mitral, en ausencia de endocarditis previa, pero la National Marfan Foundation73 sí la aconseja en pacientes con Marfan. En ausencia de más datos, parece recomendable su uso en estos pacientes.
- •
Restricción del ejercicio. Para las recomendaciones son útiles las guías publicadas por la National Marfan Fundation (http://www.marfan.org) y la Asociación Americana de Cardiología, ya indicada en el texto previo.
- •
Embarazo. Debe realizarse previamente consejo genético e información sobre riesgo obstétrico, materno y fetal. Es posible el diagnóstico preimplantacional si se detecta anomalías genéticas típicas o familiares. Debe evitarse el embarazo en mujeres con dilatación de la raíz aórtica >40mm).
- •
Evaluación oftalmológica anual. Evaluación en niños con corrección de la refracción precoz para evitar la ambliopía. Indicación de extracción del cristalino en caso de opacificación con disminución de la agudeza visual, anisometropía, error de refracción sin posibilidad de corrección quirúrgica, luxación completa o inducción de glaucoma o uveítis.
- •
Las manifestaciones esqueléticas deben tratarse según la práctica habitual. La escoliosis debe tratarse de forma quirúrgica cuando la curva excede los 40°. La artropatía asociada a hiperlaxitud puede requerir corrección quirúrgica.
- •
Guías de manejo de las enfermedades relacionadas:
- –
Evaluación cardiovascular y oftalmológica anual en fenotipo MASS, prolapso de válvula mitral sindrómico y Ehler Danlos (EL). Monitorizar el tamaño aórtico y el grado de regurgitación mitral.
- –
Consejo genético en EL o fenotipo MASS (por riesgo de presentación más severa).
- –
Evaluación cuidadosa cardiovascular y oftalmológica en niños con EM emergente o enfermedad del tejido conectivo inespecífica (tabla 3).
- –
En todos los pacientes se debe realizar desde la primera visita revisión de la historia previa y entrega de informe clínico completo con resumen de su historia anterior y actual, tratamiento y recomendaciones. En la tabla 5 se propone un protocolo de seguimiento clínico en pacientes con EM.
Protocolo de evaluación sugerido en pacientes con Marfan
| Evaluación en la primera consulta y recomendaciones iniciales: |
| 1. Evaluación diagnóstica inicial, aplicar los criterios de Ghent y/o los criterios de Ghent modificados hasta su validación. Historia clínica y exploración física completa. |
| 2. Dar información verbal y escrita personalizada de síndrome de Marfan y recomendaciones, incluidos los ajustes en el estilo de vida (reducir el riesgo de daño a la aorta, como evitar deportes de contacto o ejercicio muy esforzado). |
| 3. Realizar consejo genético, árbol genealógico y recomendación de estudio familiar. |
| 4. Inicio de tratamiento con betabloqueadores: |
| — Propanolol para conseguir FC 110 lpm en ejercicio. Se puede mejorar la cumplimentación con atenolol. En el embarazo, utilizar metoprolol o labetalol. |
| — Si hay contraindicación de betabloqueadores o precisa control adicional tensional, se sugiere losartán o incluso antagonistas del calcio (preferentemente verapamilo). |
| 5. Control estricto de los factores de riesgo cardiovascular. |
| 6. Profilaxis de endocarditis si estuviera indicada. |
| 7. Exploraciones complementarias iniciales: |
| — Analítica sanguínea, electrocardiograma y ecocardiografía. |
| — Examen ocular completo: examen del interior del ojo llevado a cabo por un oftalmólogo y utilizando lámpara de hendidura. |
| — RM/TC de la columna lumbar para descartar ectasia dural. |
| — Radiografía de tórax, de columna y de caderas. |
| Exploraciones complementarias evolutivas y recomendaciones: |
| 1. Estudio genético. |
| 2. Ecocardiograma anual en adultos con Marfan y síndrome de Marfan potencial hasta los 20 años y cada 6 meses si >45mm. Si el diámetro es normal durante exámenes consecutivos, se puede realizar cada 2 años. |
| 3. Examen de aorta completa al menos una vez con TC/RM y de forma regular si se ha realizado cirugía aortica; en este caso también se deben explorar troncos supraaórticos. |
| 4. Examen ocular periódico anual/bianual, según sintomatología. |
| 5. Controles semestrales/anuales en consulta monográfica para la vigilancia de factores riesgo cardiovascular, recopilación de exploraciones y consejo. |
No existe ningún conflicto de intereses.