




INTRODUCCION
El cáncer de mama (CM) es la neoplasia más frecuente en la mujer, con una probabilidad cercana al 5 % de desarrollar la enfermedad antes de los 75 años. El CM constituye la primera causa de muerte entre las mujeres de 35 a 64 años de edad. El riesgo de padecer cáncer de mama aumenta con la edad y llega hasta el 8 % a los 85 años.
Uno de los principales factores de riesgo es la presencia de cáncer de mama en familiares directos. Entre un 5 % y un 10 % de todos los casos presentan un componente hereditario y el porcentaje se incrementa hasta un 25 % en las mujeres menores de 35 años. Teniendo en cuenta la alta frecuencia de esta neoplasia, el cáncer de mama hereditario afecta a un gran número de mujeres en nuestra población.
Las mutaciones germinales en los genes BRCA1 y BRCA2 parecen causar la mitad de los síndromes de cáncer de mama familiar, mientras que las de BRCA1 se asocian a la mayoría de síndromes con cáncer de mama y de ovario a la vez. El riesgo estimado de padecer cáncer de mama en portadoras de una mutación en BRCA1 o BRCA2 es de un 50 % a los 50 años y superior al 80 % a lo largo de la vida. Las estimaciones para el cáncer de ovario en relación con la presencia de mutaciones en el gen BRCA1 superan el 20 % y el 60 %, respectivamente. La presencia de mutaciones en el gen BRCA2 se asocia a un riesgo menor de cáncer de ovario, pero aumenta el de cáncer de mama masculino. Ambos genes incrementan ligeramente la frecuencia de otros tipos de neoplasias (próstata, colon o páncreas) en estas familias.
IDENTIFICACION DE LOS GENESBRCA1 Y BRCA2
El análisis genético de cromosomas recombinantes de miembros de familias de alto riesgo de cáncer de mama y cáncer de ovario permitió la identificación en 17q de una región de 2 megabases, posteriormente limitada a 600 kb1, con numerosas secuencias candidatas. El estudio de su estructura genómica, patrón de expresión, características de los transcritos y, en especial, del DNA presente en individuos de familias con cáncer de mama y ovario ligados a 17q, condujo a la identificación de una secuencia que comprendía el marcador D17S855 y presentaba las características estructurales propias de un gen, que correspondía al gen BRCA12.
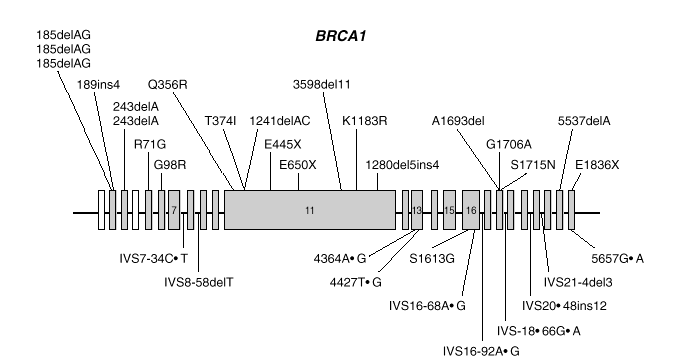
BRCA1 es un gen de gran tamaño (5.592 nucleótidos) estructurado en 24 exones (2 de ellos no traducibles). El 60 % de la secuencia corresponde a un solo exón (exón 11) y el resto se reparte en 21 exones relativamente cortos (fig. 1). El gen se extiende a lo largo de una región de 100 kb de DNA genómico, con una alta densidad de secuencias repetidas Alu, que lo hacen relativamente inestable y propenso a deleciones y reordenamientos.
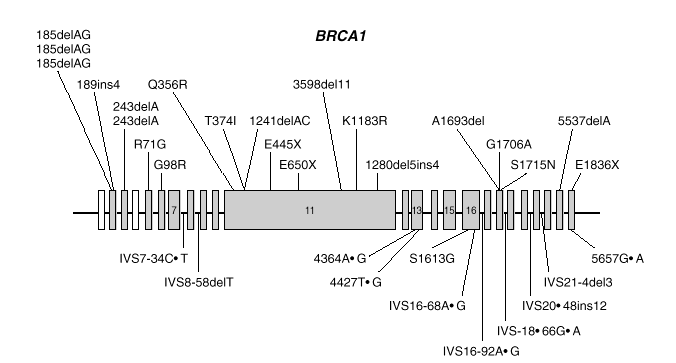
Fig. 1. Esquema del gen BRCA1 en el que las barras representan los distintos exones. En la parte superior se muestran las mutaciones y en la parte inferior los polimorfismos y variantes raras identificados por los autores en población española.
La molécula de RNAm, de 7,8 kb, se traduce a una proteína de 1.863 aminoácidos, con un peso molecular de unos 2,2 kDa. El transcrito es abundante en testículo y timo y está también presente en mama y ovario. Los primeros análisis mostraban hibridación en tejidos de hombre, ratón, rata, conejo, cordero y cerdo, pero no en pollo, indicando la conservación de BRCA1 en mamíferos.
La confirmación de BRCA1 como un gen causante de enfermedad se obtuvo identificando mutaciones patológicas en miembros de familias con susceptibilidad al cáncer de mama y ovario ligada al cromosoma 17q. Estos individuos eran portadores de mutaciones, causantes de una determinación prematura de la traducción, y del haplotipo de riesgo, lo que indicaba la cosegregación de las mutaciones con el correspondiente alelo de susceptibilidad y la enfermedad. Paralelamente, no se identificó ninguna alteración en BRCA1 en los miembros sanos de las familias.
Tras la localización del gen BRCA1 en 17q12-21 en el año 1994, el análisis de numerosas familias de alto riesgo permitió descartar su participación en un porcentaje importante de las familias con cáncer de mama3 y en las familias con cáncer de mama masculino4, sugiriendo la existencia de un segundo gen de susceptibilidad al cáncer de mama.
La búsqueda genómica en 15 familias sin ligamiento a BRCA1, permitió localizar el gen BRCA2 en la región cromosómica 13q12-135. El análisis de marcadores microsatélites adicionales en esta zona definió un espacio de 60 kb, con ligamento máximo alrededor del marcador D13S260. Posteriormente, una deleción somática de 300 kb en homozigosis encontrada en un tumor pancreático, restringió drásticamente la región candidata. El análisis, en pacientes afectados, de las secuencias de DNA contenidas en esta región condujo a la detección de una mutación, que interrumpía la traducción de la proteína resultante. La identificación de otras mutaciones que conducían a terminaciones prematuras de las síntesis proteica en familias con cáncer de mama, confirmaba la identificación del gen BRCA2, en el año 1995.
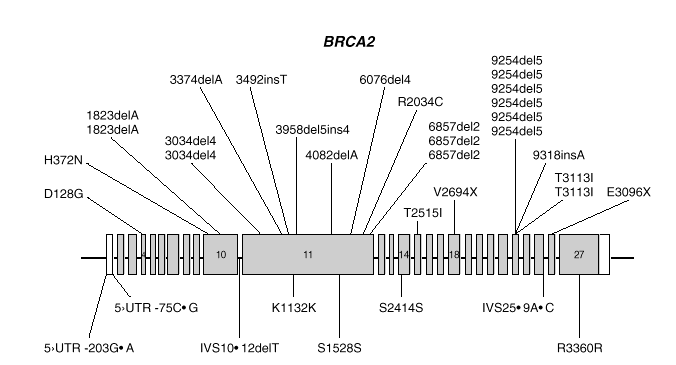
BRCA2 es, también, un gen de gran tamaño. Posee 11.385 nucleótidos, distribuidos a lo largo de unas 70 kb de DNA genómico y está compuesto de 27 exones, el primero de los cuales no se traduce. Los exones 10 y 11, de unos 1.000 y 5.000 nucleótidos respectivamente, constituyen el 60 % de la secuencia traducible. Los restantes son relativamente cortos (fig. 2). El análisis por Northern demostró un transcrito de 10-12 kb presente en células de epitelio mamario normal y tumoral, así como de placenta. La proteína sintetizada tiene 3418 aminoácidos (aproximadamente 384 kDa) y no presenta homología significativa con BRCA1 o cualquier otra proteína conocida, excepto un motivo de tipo granina situado en la zona carboxiterminal.
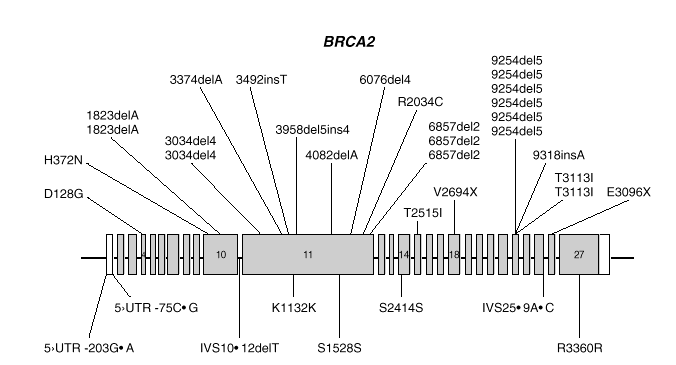
Fig. 2. Esquema del gen BRCA2 en el que las barras representan los distintos exones. En la parte superior se muestran las mutaciones y en la parte inferior los polimorfismos y variantes raras identificados por los autores en población española.
MUTACIONES EN LOS GENESBRCA1 Y BRCA2
Las mutaciones en los genes BRCA1 y BRCA2 se distribuyen uniformemente a lo largo de los dos genes6 y exceptuando un pequeño grupo de mutaciones recurrentes, las restantes son específicas de cada familia. Las figuras 1 y 2 muestran las mutaciones identificadas por los autores en población española7-9. Mayoritariamente, consisten en inserciones, deleciones o sustituciones de una o pocas bases, que provocan la alteración de la pauta de lectura causando la aparición prematura de un codón de terminación de la traducción. Las sustituciones de una base también pueden causar un cambio de aminoácido, sin alterar la longitud de la proteína. También se dan, aunque con menor frecuencia, variantes de splicing y mutaciones capaces de alterar la transcripción, así como grandes deleciones o duplicaciones. El banco de datos internacional Breast Cancer Information Core (BIC)10 recoge los centenares de mutaciones descritas hasta la fecha, la mayoría en familias con múltiples casos de cáncer de mama y ovario.
Los cambios en el DNA que causan la síntesis de una proteína truncada o anómala se consideran como mutaciones asociadas al desarrollo de un CM. La evaluación del riesgo genético es más difícil cuando se trata de sustituciones de una base nucleotídica que provoca el cambio de un aminoácido. Su asociación con la enfermedad sólo se ha podido establecer en los casos en que se ha demostrado que la mutación cosegrega con la enfermedad en mujeres afectadas de familias de alto riesgo11. Las restantes mutaciones de este tipo, se clasifican como "variantes raras" o "con efecto desconocido". Hasta que no sea posible realizar estudios funcionales de la proteína mutada, sólo es posible considerar como mutaciones patológicas, aquellos cambios que: a) no aparecen en un grupo de individuos control; b) cosegregan con la enfermedad en una familia; c) causan la sustitución de un aminoácido por otro que posee unas características muy diferentes; d) afectan a aminoácidos muy conservados a lo largo de la evolución, y e) se localizan dominios potencialmente funcionales de la proteína.
Se ha detectado en diversas familias, con ligamiento al BRCA1, la ausencia de transcripción del alelo asociado al cáncer. Se ha sugerido12 que entre un 15 % y un 20 % de las mutaciones afecta a la expresión, al splicing o a la estabilidad del transcrito. Se ha documentado, también, la existencia de deleciones de gran tamaño que originan la pérdida de algún exón o de la región promotora del gen13, como resultado de la recombinación no homóloga entre secuencias Alu, así como duplicaciones de fragmentos que contienen exones enteros. En un porcentaje considerable de familias con ligamento, en las que no se han identificado alteraciones genéticas mediante las técnicas más comúnmente usadas para la búsqueda de mutaciones (SSCP, PTT, secuenciación directa), pueden presentarse grandes alteraciones de la secuencia del gen que son detectables mediante el uso de metodologías adicionales como el Southern o la PCR inversa.
INTERPRETACION DEL ANALISIS GENÉTICO
Con la clonación de los genes BRCA1 y BRCA2 se pueden ofrecer pruebas predictivas a miembros de familias a riesgo, aunque, a diferencia de otros genes asociados a cáncer (APC, RB1, RET), la conveniencia de su aplicación y las implicaciones de un resultado positivo o negativo, no son siempre claras.
En el cáncer de mama y ovario hereditarios el análisis genético está justificado en familias de alto riesgo, en las cuales permite estimar más correctamente el riesgo de aparición del cáncer. Este conocimiento es esencial en el momento de tomar decisiones clínicas. En algunas circunstancias pueden aplicarse los análisis genéticos al cribaje de aquellas poblaciones con un número limitado de mutaciones debidas, algunas de ellas a la existencia de un efecto fundador, aunque el beneficio real en términos económicos o humanos es todavía desconocido.
En familias pequeñas en que la probabilidad de poseer una mutación es baja, la aplicación de análisis genéticos puede generar expectativas irreales que incrementen la ansiedad, ya que probablemente, no sean concluyentes. Un resultado negativo puede interpretarse erróneamente como la ausencia de riesgo y llevar al abandono de las medidas o programas de detección precoz de estas neoplasias. Alternativamente, los portadores de mutaciones en estos genes, pueden llegar a tomar decisiones clínicas con un beneficio para su salud discutible, dada la incerteza de las estimaciones del riesgo asociado a estas mutaciones.
La interpretación correcta de los resultados individuales constituye una de las principales dificultades de la aplicación de las pruebas genéticas. La no detección de mutación en BRCA1 y BRCA2 no es un resultado de absoluta certeza, exceptuando cuando se ha identificado una mutación en otros miembros de la familia. En este caso, el sujeto no comparte el riesgo familiar a padecer cáncer de mama, pero sí comparte el riego de la población general. Deben tenerse en cuenta otros factores a la hora de interpretar un resultado negativo: a) puede haber mutaciones en BRCA1 o BRCA2 no detectables mediante la metodología utilizada o disponible (más del 20 % según el BCLC); b) no todos los cánceres de mama heredados son debidos a los genes BRCA1 o BRCA214 y es posible que se identifiquen, en un futuro no muy lejano, otros genes de susceptibilidad; c) puede haberse heredado una mutación en otro gen que no produzca, como efecto primario, cáncer de mama, pero sí un incremento en el riesgo de sufrirlo, o puede tratarse de genes recesivos o con una baja penetrancia, que contribuyen en menor grado el riesgo de sufrir cáncer de mama, como los genes ATM y PTEN, entre otros, y d) puede ocurrir que una agrupación familiar de cáncer de mama y ovario haya ocurrido al azar dada la alta prevalencia del cáncer de mama esporádico.
Frente a un resultado positivo, no siempre es fácil determinar el riesgo asociado a una determinada variante genética. La sustitución de una base puede ser desde un polimorfismo neutro a una mutación altamente patógena15. Pero, aunque el efecto deletéreo sea claro, la evaluación exacta del riesgo individual no es sencilla. La existencia de genes modificadores, una historia reproductiva, o factores ambientales, entre otros, alteran los efectos de las mutaciones heredadas en genes de predisposición en cada individuo. Considerando, además, que una misma mutación en distintas pacientes puede asociarse a distintos grados de riesgo o a diferentes tipos de cáncer, hay que proceder con las máximas precauciones al transmitir la información a las familias afectadas.

