






Introducción
El síndrome de Kabuki, también conocido como síndrome de maquillaje Kabuki (Kabuki make-up), fue descrito por primera vez de manera simultánea en 1981 por los autores japoneses Niikawa y colaboradores (1) y Kuroki y colaboradores (2).
El término kabuki significa cantar (ka), bailar (bu), y habilidad (ki). Frecuentemente se traduce kabuki como "el arte de cantar y bailar". La denominación de este síndrome, dada por Kiikawa en su trabajo, se debe al parecido de estos niños con el maquillaje de los actores del teatro tradicional japonés Kabuki, especialmente, por el aspecto de la eversión del borde inferior externo de los párpados.
Este síndrome fue descrito inicialmente en personas de origen japonés, si bien es cierto es el lugar donde más comúnmente se presenta, con una frecuencia estimada de 1/32,000 recién nacidos vivos y cada vez se diagnostican más casos tanto dentro como fuera de Japón (3). Se informa en la bibliografía sobre dos series en España: una con de 18 casos (4) y otra con cinco (5); un informe en Arabia saudita y algunos otros casos esporádicos en otras regiones del mundo como Australia, Ecuador, Brasil, el norte de Europa y Nueva Zelanda (3). Sólo encontramos un caso mexicano informado en 2006 (6).
El propósito de este artículo es presentar el caso clínico de un niño con diagnóstico de síndrome de Kabuki en México. Los hallazgos oftalmológicos son parte fundamental para establecer el diagnóstico ya que genéticamente no se dispone de un estudio específico para su confirmación diagnóstica, por lo que sigue siendo un diagnóstico basado en la clínica, de la cual las manifestaciones oftalmológicas se presentan en 100% de los casos informados.
Informe del caso
Se trata de un niño de cuatro años de edad, producto de la quinta gesta, que presentó amenaza de parto prematuro a los cinco meses de gestación, resuelta con reposo; obtenido por parto eutócico, de término, dado de alta junto con la madre. Se desconoce su Apgar. Madre de 28 años y padre de 39 años, sanos, no consanguíneos, tiene tres hermanos sanos. A los dos días de vida extrauterina, se le diagnosticó coartación de la aorta por lo que fue sometido a procedimiento de balón dilatado, sin complicaciones en la cirugía. En terapia intensiva presentó neumonía por lo que estuvo hospitalizado por dos meses. Se realizó ultrasonografía (USG) de vías urinarias donde se detectó agenesia renal y criptorquidia bilateral. Presenta también quiste de erupción en paladar superior, que requirió de cirugía maxilofacial. A los dos años siete meses se le realizó orquidopexia bilateral. Presenta retraso psicomotor moderado. Por todo lo anterior y su facies característica (Figura 1), se le diagnosticó portador del síndrome de Kabuki a los tres años seis meses por el servicio de genética. Se le realizó cariotipo encontrándose normal: 46, XY.
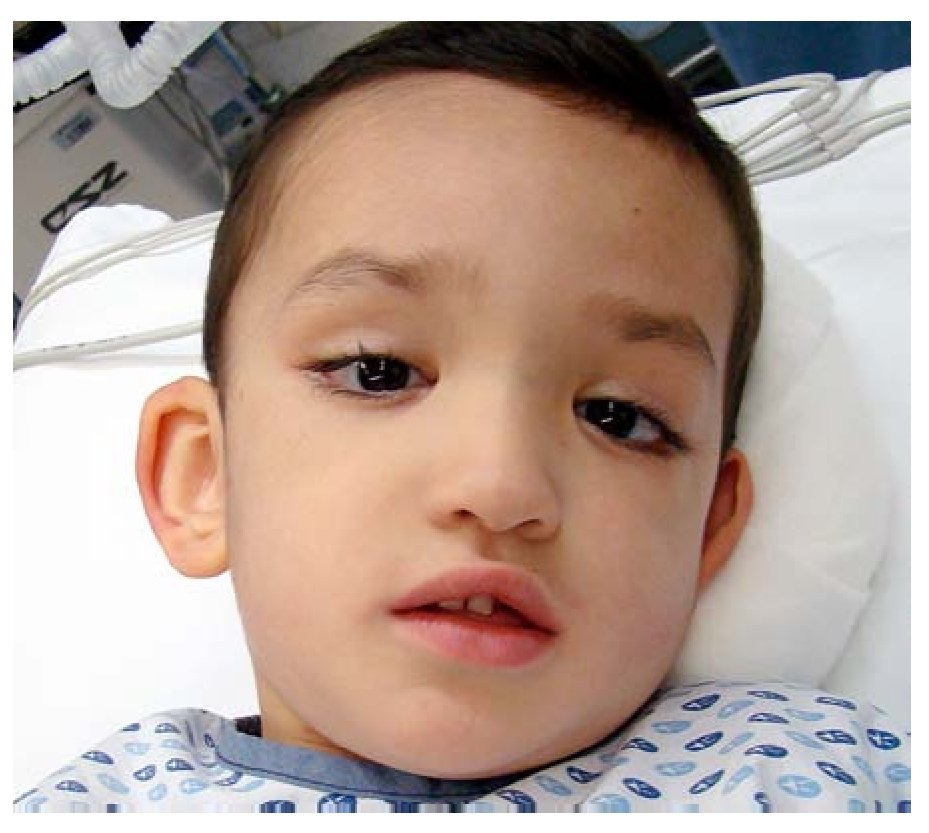
Figura 1. Facies características. Se aprecia la raíz nasal ancha, la punta nasal rectificada, las alas nasales levemente hipoplásicas, la boca pequeña, los pabellones auriculares grandes, así como el aspecto oriental característico de estos pacientes. Nótese la endotropia y el euriblefarón.
El examen físico muestra telecanto, eversión del tercio lateral de los párpados inferiores con pliegue palpebral elongado (euriblefarón), cejas arqueadas con tercio lateral más despoblado (Figura 2), raíz nasal ancha con punta deprimida y alas levemente hipoplásicas, boca pequeña con labio superior en V invertida, paladar ojival, micrognatia y pabellones auriculares grandes y anormalmente plegados, implantación del cabello baja tanto en región frontal como occipital, almohadilla en la punta de los dedos e incremento del asa cubital e hipotenar (Figura 3).

Figura 2. Eversión del tercio externo palpebral inferior (euriblefarón).
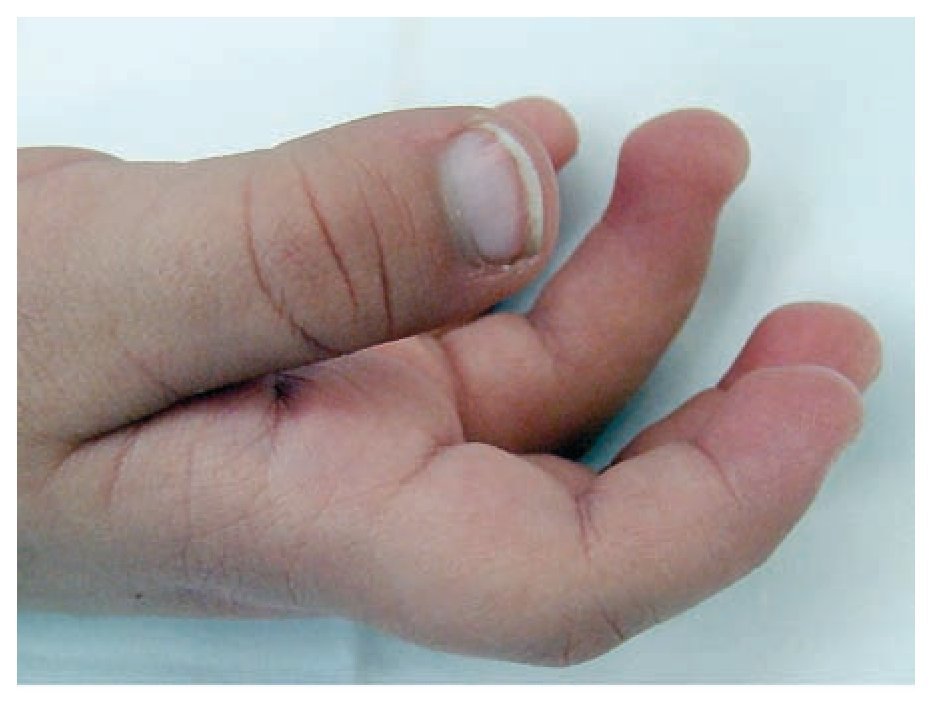
Figura 3. Manos pequeñas. Almohadillas en los pulpejos.
Al examen oftalmológico presentó, además de lo anterior, síndrome del parpadeo mandibular de Marcus Gunn, detectado desde los primeros días de vida, posteriormente a los tres años se diagnostica ambliopía de ojo izquierdo que se trató con oclusión dos horas al día, además de una endotropia de 30 D con una hiperfunción de oblicuo inferior derecho 2+ y del izquierdo de 1+. Se decidió realizar corrección de estrabismo a los cuatro años y un mes de edad mediante un retroimplante del músculo recto medio de ojo izquierdo y resección del recto lateral del mismo ojo, con una miectomía del oblicuo inferior del ojo derecho y un procedimiento en "z" del oblicuo inferior del ojo izquierdo, mostrando buena evolución (Figura 4). Por lo demás es oftalmológicamente sano. Está pendiente corregir la eversión del párpado inferior.
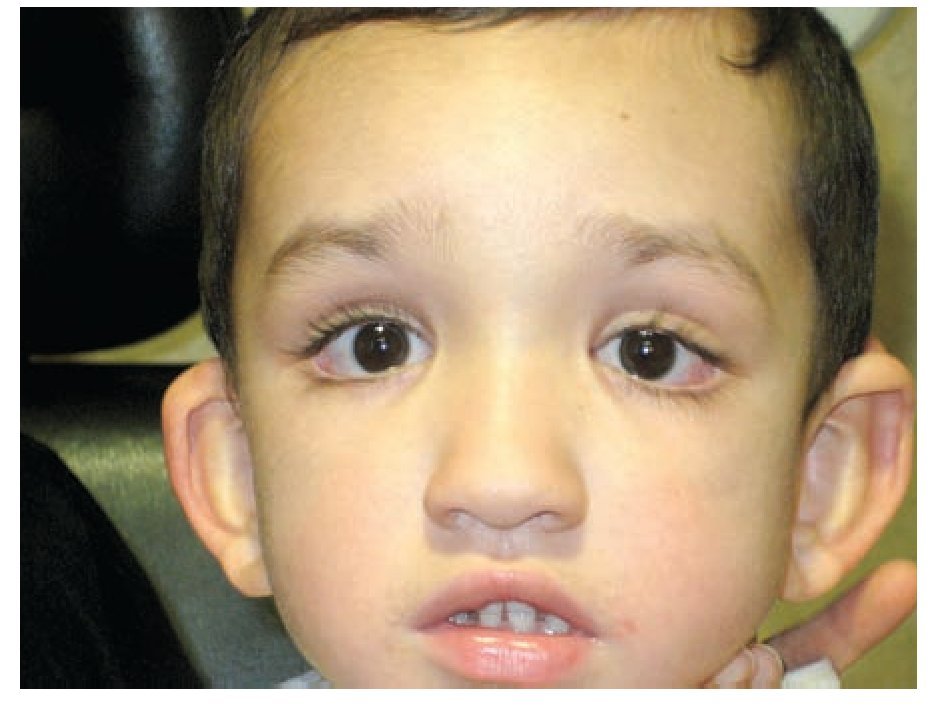
Figura 4. Estado posoperatorio por estrabismo, a un mes de evolución.
Discusión
El síndrome Kabuki es una entidad poco frecuente que se caracteriza por múltiples anomalías congénitas y retraso mental. Sus signos faciales típicos están presentes desde momentos tempranos de la vida y son cruciales para su diagnóstico.
La causa del síndrome Kabuki se mantiene desconocida. Se han descrito familias con patrones de herencia autosómica dominante, recesiva ligada al cromosoma X e incluso se ha asociado a numerosas anomalías citogenéticas. Estos hallazgos de laboratorio, junto a la naturaleza multisistémica de esta afección, sugieren que el síndrome Kabuki pudiera estar causado por una microduplicación que afecta varios genes o por un gen que regula varios genes "blancos" o claves (7).
Existe en la actualidad una red de este síndrome, conocida como "La Red del Síndrome Kabuki" (RSK), y puede decirse que ha sido contactada ya por más de 180 familias. La RSK en Holanda tiene cerca de 20 miembros en un área geográfica pequeña (8). El Dr. Niikawa ha documentado al menos 100 casos en Japón y una genetista de Brasil ha reportado al menos 15 familias afectadas en su país (9).
En un estudio publicado en noviembre del 2003 por el Dr. Milunsky y colaboradores descubrieron una duplicación submicroscópica en el cromosoma 8, porción 8p 22-23 (10).
Lo cierto es que hasta la fecha se desconoce la etiología de este síndrome, la mayoría de los casos descritos son de tipo esporádico sin predilección de sexo, ni relación con la edad materna. En algunos pacientes el problema es hereditario autosómico dominante de expresión variable (7).
La supervivencia suele ser normal. Los primeros casos descritos tienen actualmente 30 años de edad. Todavía son pocos los casos conocidos fuera de Japón, encontramos casos esporádicos informados en América del Norte, Brasil, Filipinas, Vietnam, Arabia, India y África (11).
En Latinoamérica, el mayor número de casos comunicados son de Brasil (9), existe uno reportado en Haití (10), uno en Ecuador (12) algunos casos esporádicos en Cuba (13,14) y hasta ahora uno en México (6). La prevalencia es cada vez mayor fuera de Japón, lo que indica que el número de pacientes no diagnosticados puede ser alto, debido a no estar tan difundido el conocimiento de este síndrome.
Al no contar con un estudio específico para su diagnóstico, la clínica es lo que nos debe hacer sospechar. Diversos autores han señalado las características fundamentales que presentan estos pacientes. El estudio más representativo acerca de esto es el descrito por Niikawa y colaboradores (3) en 1988 donde, a partir de 62 pacientes, sentaron las bases del diagnóstico de este síndrome sobre cinco manifestaciones cardinales:
1. Características faciales peculiares, como nariz con punta ancha y deprimida, cejas arqueadas, eversión del tercio externo del párpado inferior y orejas grandes y de implantación baja (100%).
2. Anormalidades esqueléticas, como deformidades en columna vertebral y braquidactilia (92%).
3. Anormalidades de los dermatoglifos, almohadilla en la punta de los dedos, incremento del asa cubital e hipotenar (93%).
4. Retraso psicomotor moderado (92%).
5. Talla baja (83%).
Estos criterios siguen siendo vigentes. Otro estudio más reciente, realizado por Adam y colaboradores (11), informaron una serie de 10 pacientes japoneses con síndrome de Kabuki, seguidos en un periodo de 16 años, con los mismos cinco rubros diagnósticos como criterios mayores y algunas características menos comunes, como criterios menores, tales como anormalidades viscerales, desarrollo prematuro de caracteres sexuales secundarios en las niñas y susceptibilidad a infecciones.
Específicamente las manifestaciones oftalmológicas encontradas en todos los pacientes son cejas arqueadas, pestañas largas, eversión del tercio externo del párpado inferior (euriblefarón) y elongación de la fisura palpebral horizontal.
Encontramos en la literatura otras manifestaciones oftalmológicas menos frecuentes como microftalmía, coloboma (15), miopía, ptosis, estrabismo, (en especial endotropias), escleróticas azules, opacidades corneales (16). En una revisión realizada por Matsumoto y colaboradores (17) se encontraron las siguientes manifestaciones oftalmológicas como criterios mayores: fisura palpebral elongada (99%), cejas arqueadas (85%), ptosis (50%). Dentro de los criterios menores se encontró escleras azules (31%) y coloboma de párpado (27%) (13). Otro artículo reporta el caso de un paciente con Kabuki y Jaw-Winking o ptosis de Marcus Gunn (18) que, hasta su publicación, no había sido mencionada esta asociación, y resulta interesante que nuestro paciente también la presentó.
Elsherbiny (19) comunicó un caso con depósitos maculares amarillos no asociados a edema en un niño de seis años, como otro hallazgo oftalmológico en el síndrome de Kabuki, que puede prestarse a hacer diagnóstico diferencial con el raro síndrome de Hardikar, que presenta características comunes al síndrome de Kabuki, y se caracteriza por estenosis/atresia congénita de las vías biliares intrahepáticas y distrofia retiniana. Esta asociación también fue reportada por Cols y Haeken en una antigua publicación, en 1997 (20). Por lo anterior creemos de gran importancia estudiar a fondo cualquier paciente con alguna de estas características para descartar el síndrome de Kabuki, así como el envío a valoración oftalmológica en todos los casos ya diagnosticados.
Hasta la fecha no se han encontrado que estas manifestaciones sean secundarias a otras condiciones patológicas coincidentes. Es probable que formen parte de esta entidad y sean de aparición tardía. Aunque se plantea que en esencia la expresión clínica del síndrome Kabuki sea similar a lo largo de toda la vida, el hecho de que la mayoría de los casos hayan sido descritos en la bibliografía en edades pediátricas, limita el conocer datos relativos a su evolución y el hecho de que fue descrito en 1981; los casos más antiguos tienen alrededor de los 30 años, por lo que es de suma importancia el seguimiento de estos pacientes.
Conclusiones
La mayoría de los casos publicados se han diagnosticado en la infancia tardía y preadolescencia. Dada la mayor difusión de este síndrome cada vez se diagnostican más tempranamente. Los rasgos faciales característicos presentes desde las primeras etapas de la vida, junto con el retraso del crecimiento y retraso del desarrollo psicomotriz, permiten establecer precozmente la sospecha diagnóstica. Por lo anterior es importante fomentar el conocimiento de este síndrome, no sólo entre los distintos especialistas pediátricos, sino también entre los pediatras generales, oftalmólogos y otros profesionales de la salud, para posibilitar un diagnóstico temprano y así poder ofrecer una mejor expectativa de vida.
Correspondencia: Dra. Brenda Ivette Trigueros Becerra.
Instituto de Oftalmología, S.C. Rio San Juan No. 103 (1 piso) Col. Miravalle, Monterrey, N.L. CP 64660.
Tels. (81) 8356-1878 y 1884, Celular. 81166 09522, Fax. (81) 8356 17 99,
Correo electrónico: bitrigueros@hotmail.com
