













Introducción: La coriorretinopatía en perdigonada (birdshot) es una enfermedad autoinmune rara, que se observa con mayor frecuencia en personas de procedencia europea. En nuestro grupo étnico, se conoce muy poco de su prevalencia y espectro clínico. Con esta serie de casos, pretendemos mostrar las características clínicas, complicaciones y manejo terapéutico de la enfermedad, en nuestra población.
Serie de casos: Presentamos una serie de seis pacientes con coriorretinopatía en perdigonada, en diferentes etapas del curso clínico de la enfermedad, que acudieron al servicio de inmunología y uveítis de nuestra Institución. Se describen la presentación, características clínicas, complicaciones, manejo terapéutico y resultado del mismo, en cada caso.
Discusión: Tres hombres y tres mujeres, con una edad promedio de 46.5 años, todos de origen mexicano. Los seis pacientes tenían lesiones coriorretinianas típicas, redondas u ovales, amarillo-cremoso y de distribución peripapilar "en perdigonada". Todos ellos presentaron un fondo vitiliginoso y cierto grado de vitreítis y vasculitis retiniana. En los cinco pacientes, que se realizaron la determinación del haplotipo HLA-A29, el resultado fue positivo. Sólo dos de seis pacientes acudieron a nuestra clínica al inicio de su padecimiento, los cuatro restantes habían sido tratados previamente por uveítis de origen desconocido en otro hospital. Estos cuatro pacientes presentaron complicaciones oculares, que derivaron en pérdida visual significativa por retraso en el diagnóstico y falta de tratamiento adecuado.
Introduction: Birdshot retinochoroidopathy is a rare autoimmune disease more frequently seen in European descendents. In our ethnic group, little is known about its prevalence and clinical spectrum. The following case series is intended to show the clinical manifestations, ocular complications and management of the disease in our population.
Case series: We present herein a series of six patients with birdshot retinochoroidopathy that were seen at the Immunology and Uveitis Service of our institution at different stages of the clinical course of the disease. We describe the presentation, clinical manifestations, ocular complications, management outcome in every case.
Discussion: Three men and three women with a mean age of 46-5 years, all of Mexican origin. All patients had typical chorioretinal lesions: round or oval yellow-cream colored, of peripapillary distribution in a "birdshot" pattern. All patients had a fundus vitiliginous appearance and moderate vitritis, and retinal vasculitis. In five patients tested for the HLA-A29 haplotype, the result was positive. Only two of six patients were seen in our clinic from the beginning of the disease, the rest had been treated previously as idiopathic uveitis elsewhere. These four patients had ocular complications that resulted in significant loss of vision because of delay diagnosis and inappropriate therapy
¿ Introducción
La coriorretinopatía en perdigonada (birdshot) es una uveítis bilateral rara de origen autoinmune, con un curso clínico crónico recurrente, que afecta primordialmente el segmento posterior del ojo.
Su patología fue descrita por vez primera, en 1949 por Franceschetti y Babel, quienes la llamaron "choriorétinite en taches de bougie" (coriorretinitis en mancha color de cera).1 Treinta años después, Thomas Aaberg en 1979, la llamó coroidopatía en "parches de salmón".2 No fue sino hasta 1980, que Ryan y Maumenee describieron una serie de casos de la misma enfermedad, denominándola "retinocoroidopatía de birdshot", nombre con el cual se le conoce actualmente y adoptado por el patrón de las lesiones coriorretinianas simulando un "disparo de escopeta".3 Otros sinónimos de esta entidad fueron aportados por Gass en 1981, quién enfatizó la apariencia de las lesiones despigmentadas, llamándola "coriorretinitis vitiliginosa".4 Al mismo tiempo, en Francia, Amalric y Cuq la denominaron "chorioretinopathie en grains de riz" (coriorretinopatía "en granos de arroz").5
La coriorretinopatía en perdigonada (birdshot) es una enfermedad poco frecuente, representando del 0.6% al 1.5% de todos los casos de uveítis, remitidos a centros de alta especialidad.6,7 Existe una muy leve predilección por el género femenino (54.1% casos en general). La edad promedio de presentación es 53 años (rango entre 15 y 79 años).8-10 La gran mayoría de los pacientes descritos son de raza blanca, ascendentes del norte de Europa.4,8,9
La presentación clínica inicial se caracteriza por disminución de la agudeza visual, debida a edema macular o miodesopsias ocasionadas por vitreítis.6,10 Los pacientes afectados también pueden presentar nictalopia, discromatopsia, fotopsias y deslumbramiento.3,8 Ambos ojos se encuentran afectados, aunque el compromiso puede ser asimétrico. Algunos pacientes pueden permanecer asintomáticos o con molestias mínimas al inicio de la enfermedad, lo cual retarda el diagnóstico.6,8
Mientras que la cámara anterior muestra un mínimo o ningún grado de inflamación, en el segmento posterior, es común observar vitreítis moderada en todos los casos.8-10 Además, es frecuente observar vasculitis retiniana con afección de vasos pequeños y grandes en el polo posterior, así como hemorragias perivasculares en la capa de fibras nerviosas y tortuosidad vascular, con estrechamiento de arteriolas y vénulas.6,8-10 El edema macular quístico y la papilitis también son frecuentes.8,9 En muchos de los casos, el nervio óptico muestra una palidez progresiva y ocasionalmente puede aparecer neovascularización del disco óptico o en otra parte de la retina, provocando hemovítreo espontáneo.8,11
Las lesiones coroideas típicas que dan lugar al nombre de esta patología son hipopigmentadas, de forma redonda u oval, de color amarillo-naranja crema, y de bordes no definidos.3 Dichas lesiones tienden a seguir un patrón radial con respecto al disco óptico, localizándose principalmente en la retina nasal e inferior, con una distribución relativamente simétrica en ambos ojos.8-10 Usualmente, no sobrepasan el ecuador anteriormente y miden desde un cuarto, hasta un diámetro de disco óptico.9 Con el tiempo, las lesiones pueden confluir en grandes áreas geográficas de hipopigmentación que parecen radiar del nervio óptico, confiriendo el patrón vitiliginoso descrito por Gass.4 Estas lesiones representan la característica diagnóstica más importante de la coriorretinopatía en perdigonada, no obstante, se pueden presentar hasta varios años después de iniciado el proceso inflamatorio, retrasando el diagnóstico.12
A pesar de que el diagnóstico de la coriorretinopatía en perdigonada está basado en los hallazgos clínicos antes descritos, estudios auxiliares de diagnóstico contribuyen de manera significativa al mismo. El haplotipo HLA-A29 se encuentra presente, hasta en un 98% de pacientes con coriorretinopatía en perdigonada, mientras que sólo se encuentra en el 7% de la población general, representando hoy en día, la asociación más estrecha entre una molécula del complejo de histocompatibilidad mayor clase-I (MHC-I), con una enfermedad en humanos.13-15
En la fluorangiografía, las lesiones coroideas inicialmente aparecen hipofluorescentes y en las fases tardías, hay una leve hiperfluorescencia.16-18 Existen diversos grados de tinción y fuga fluoresceínica de capilares peripapilares y perimaculares, así como edema macular quístico y áreas retinianas de hiperfluorescencia difusa, en las fases tardías del fluorangiograma.4,9
Gass4 y colaboradores describieron que durante la fluorangiografía de retina, se observa un retraso en el tránsito arteriovenoso. Sin embrago, estudios más recientes con verde de indocianina (ICG), han demostrado que en realidad hay una exudación masiva de fluoresceína durante la fase arterial, lo cual causa insuficiencia fluoresceínica para llenar los vasos durante la fase venosa.16-18 En la ICG, el hallazgo principal es la presencia de puntos oscuros hipofluorescentes durante la fase intermedia del angiograma, los cuales permanecen hipo o isofluorescentes en las fases tardías del estudio.17 Otros dos signos frecuentes, son la pobre visualización de vasos coroideos y la hiperfluorescencia coroidea difusa tardía.17,18
Estudios electrofisiológicos demuestran elevación en el umbral de adaptación a la obscuridad y discromatopsia adquirida, en el eje azul-amarillo.19,20 El electrorretinograma (ERG) se caracteriza por una amplitud reducida, un incremento en la latencia de la onda "b" y la ausencia de potenciales oscilatorios, tanto en conos como en bastones.19 Se ha reportado que las ondas "b", se ven afectadas de forma más temprana que las ondas "a". Aunque, recientemente se ha descrito que ambas ondas se ven afectadas por igual.21 El electro-oculograma puede estar normal o subnormal.20
Finalmente, la campimetría visual automatizada confirma la depresión difusa y progresiva de la función retiniana y la presencia de escotomas variables.22,23
A pesar de que la etiología de la coriorretinopatía en perdigonada es desconocida, existe una clara asociación con el haplotipo HLA-A29. El riesgo relativo de desarrollar la enfermedad se ha estimado entre 50 y 224 veces más,14,15 la sensibilidad y especificidad diagnóstica del HLA-A29 se ha estimado en un 96% y 93%, respectivamente.24,25 Aunque en un principio se asoció la enfermedad exclusivamente al HLA-A29.2, recientemente se ha comprobado que ambos subtipos, A29.1 y A29.2 están asociados con esta patología.26,27 El mecanismo por el cual las moléculas MHC-I intervienen en el desarrollo de la enfermedad, no se conoce.
Por otro lado, la mayoría de los pacientes con coriorretinopatía en perdigonada exhiben respuestas linfoproliferativas "in vitro", contra el antígeno "S" (soluble) retiniano y contra la proteína retiniana ligadora de fotoreceptores (IRBP).13 También se ha reportado la enfermedad en mellizos monocigóticos.28 Estos hallazgos sugieren una predisposición genética y que la autoinmunidad retiniana, juega un papel crucial en su desarrollo.13,28 Actualmente, se han encontrado concentraciones elevadas de IL-17, IL-2, IL-6 y TNF-alfa en humor acuoso de pacientes con coriorretinopatía en perdigonada, reafirmando que los linfocitos-Th17 juegan un papel primordial, en el desarrollo de la enfermedad y enfatizando su carácter autoinmune.29
El estudio histopatológico de un ojo con coriorretinopatía en perdigonada en fase activa leve, mostró múltiples focos inflamatorios con predominio linfocitario a varios niveles de la coroides, llenando sus canales vasculares.30 Se observaron células plasmáticas escasas y algunos focos inflamatorios, estaban asociados a hemorragia y presencia de células epitelioides sin necrosis. No se observó afección del epitelio pigmentario de la retina (EPR), sobre las zonas de inflamación coroidea.30 En contraste a lo anterior, un estudio de imagen empleando tomografía óptica de coherencia de ultra-alta resolución (UHR-OCT), mostró atrofia de fotorreceptores en diversas áreas, así como degeneración y atrofia del EPR, bajo áreas de fotorreceptores afectados. Además, las capas internas de la retina mostraron desorganización.31
La historia natural de la enfermedad se caracteriza por la cronicidad, presentando exacerbaciones agudas y una disminución insidiosa de la agudeza visual y reducción progresiva de los campos visuales.22,23 Este curso clínico caracterizado por exacerbaciones inflamatorias vasculares, papilares y maculares que ponen en riesgo la visión del paciente, así como la potencial disfunción retiniana crónica progresiva y subclínica observada en algunos casos, han hecho que muchos expertos recomienden utilizar los estudios electrofisiológicos y/o campimétricos, para el seguimiento de los pacientes.32,33 Además, este comportamiento agresivo de la enfermedad, ha hecho que el tratamiento esté basado en terapia inmunosupresora por tiempo prolongado.34-36
Debido a la escasa información sobre la ocurrencia de esta enfermedad en nuestro país, decidimos realizar este reporte de casos clínicos, con el propósito de mostrar el espectro clínico y la experiencia terapéutica de la coriorretinopatía en perdigonada, en pacientes provenientes del noreste de México. Creemos que esta contribución es importante, ya que aunque se trata de una patología rara, es causa de pérdida visual significativa, cuando el diagnóstico no se realiza de manera oportuna y el tratamiento no es el adecuado.
¿ Reporte de casos clínicos
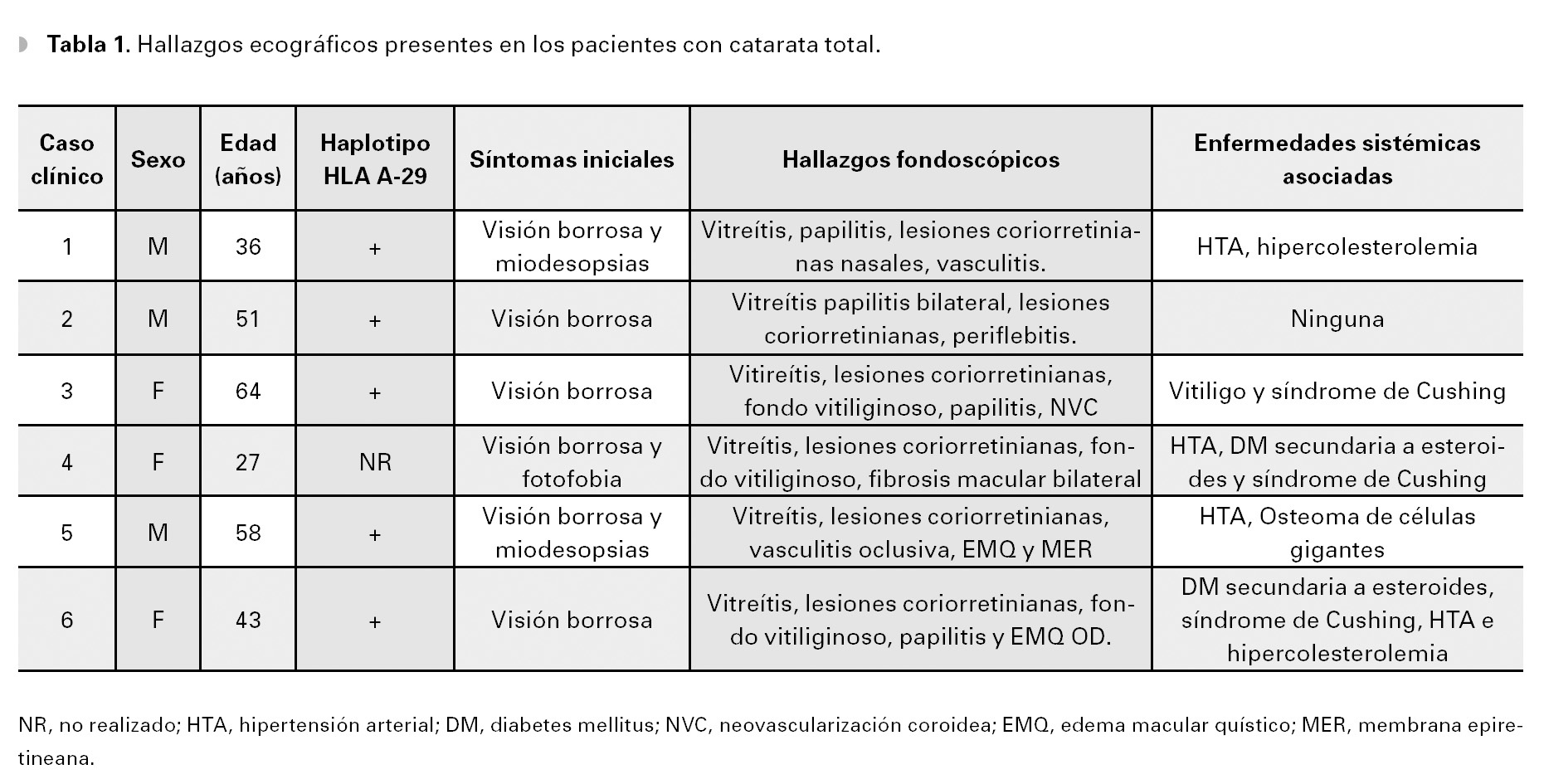
Caso 1. Masculino de 36 años de edad, que acude a consulta por visión borrosa y miodesopsias de varias semanas de evolución en ambos ojos, siendo más acentuadas en ojo derecho. El paciente tiene antecedentes de hipertensión arterial sistémica descontrolada e hipercolesterolemia.
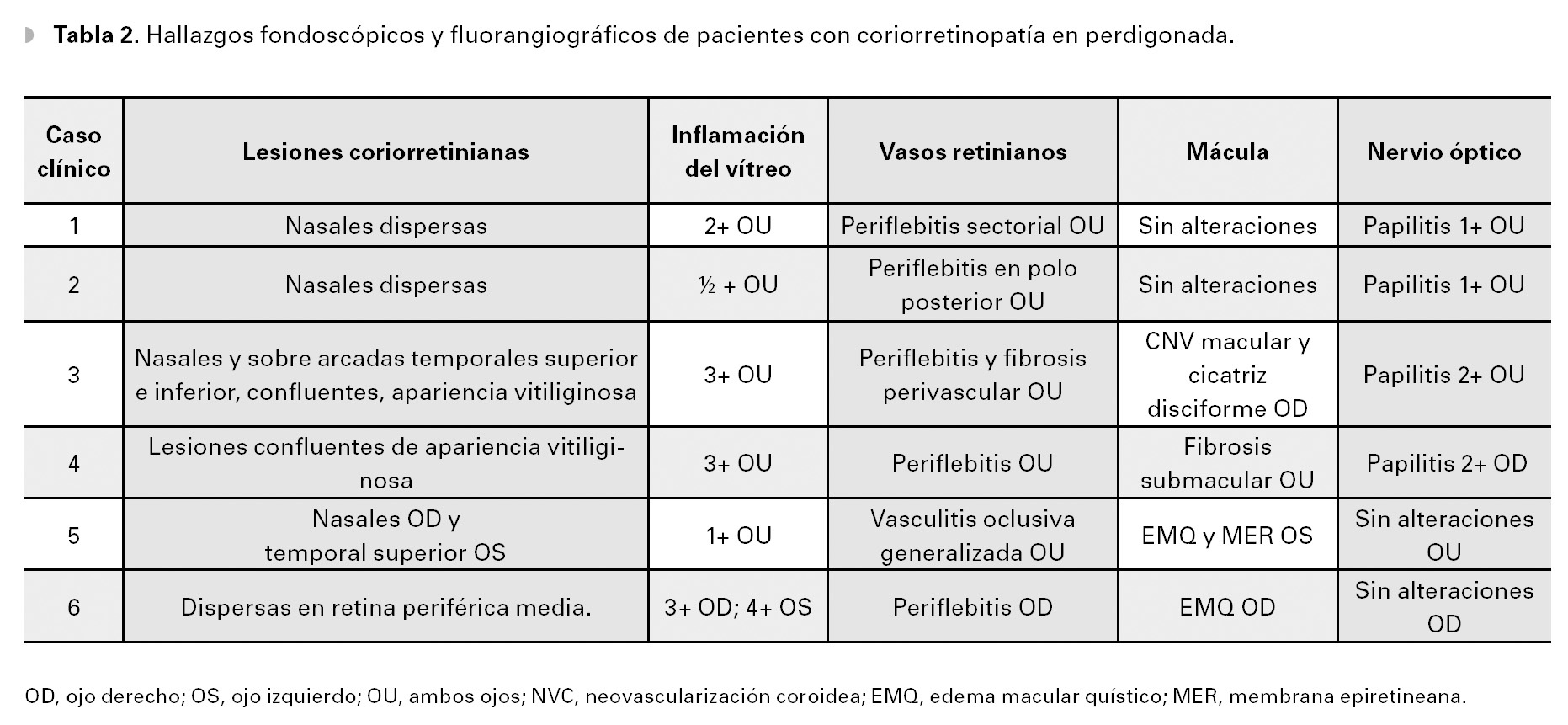
A la evaluación oftalmológica presentó una agudeza visual no corregida de 20/30 OD y 20/20 OS, así como discromatopsia caracterizada por desaturación al rojo (tablas de Ishihara). En el segmento anterior, no se observaron signos de inflamación alguna y la presión intraocular se encontró en 12 mmHg OU. La fundoscopía reveló una vitreítis moderada (2+) OU, ligero borramiento de los márgenes del disco óptico, así como una serie de lesiones ovales e hipopigmentadas a nivel profundo en la retina, de coloración amarillo cremosas y de distribución predominantemente nasal, alrededor del nervio óptico. Además, se observaron exudados inflamatorios perivasculares sectoriales en arteriolas y vénulas de ambos ojos, aunque más acentuado en ojo derecho (Figuras 1A y 1B).
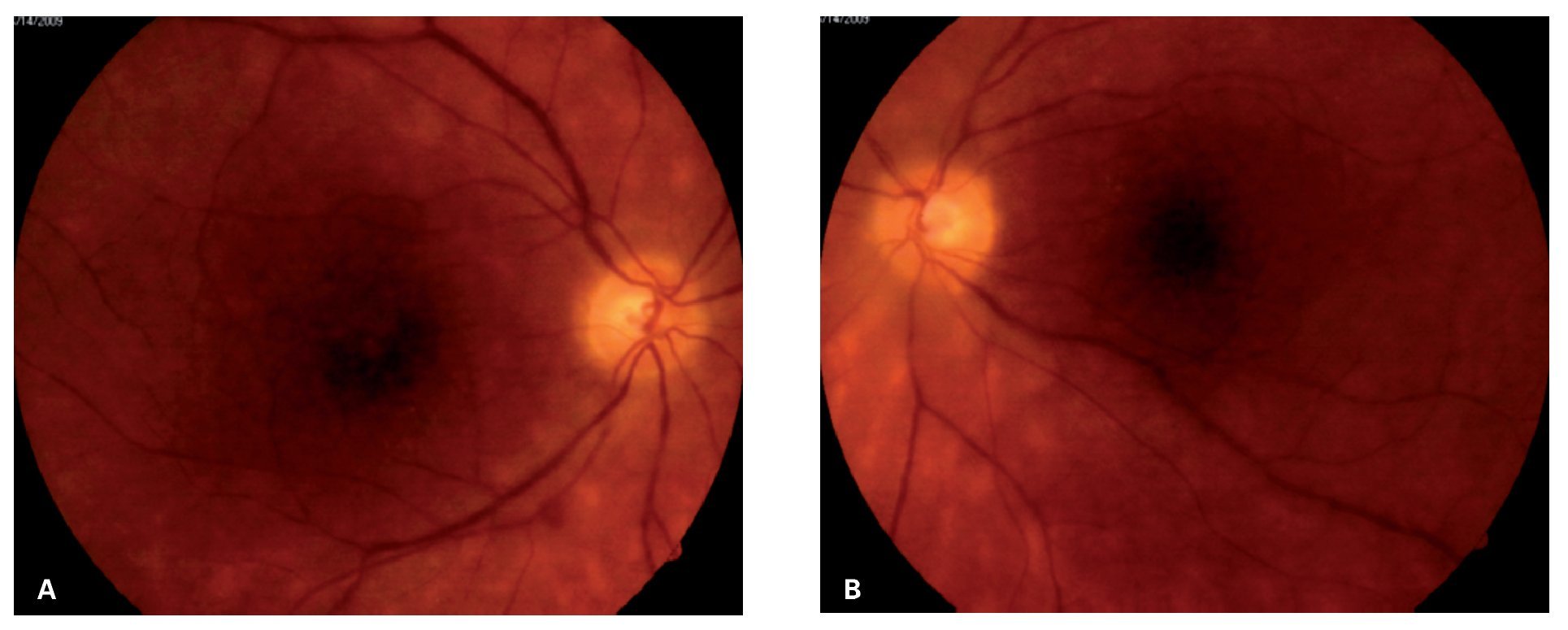
¿ Figura 1. Cuadrante nasal inferior del ojo derecho (caso 1) mostrando lesiones ovales, amarillo-cremosas, de distribución radial en retina periférica media, características de coriorretinopatía en perdigonada y envainamiento venular sectorial A). Cuadrante nasal inferior del ojo izquierdo del mismo paciente, mostrando las mismas características patológicas antes descritas B).
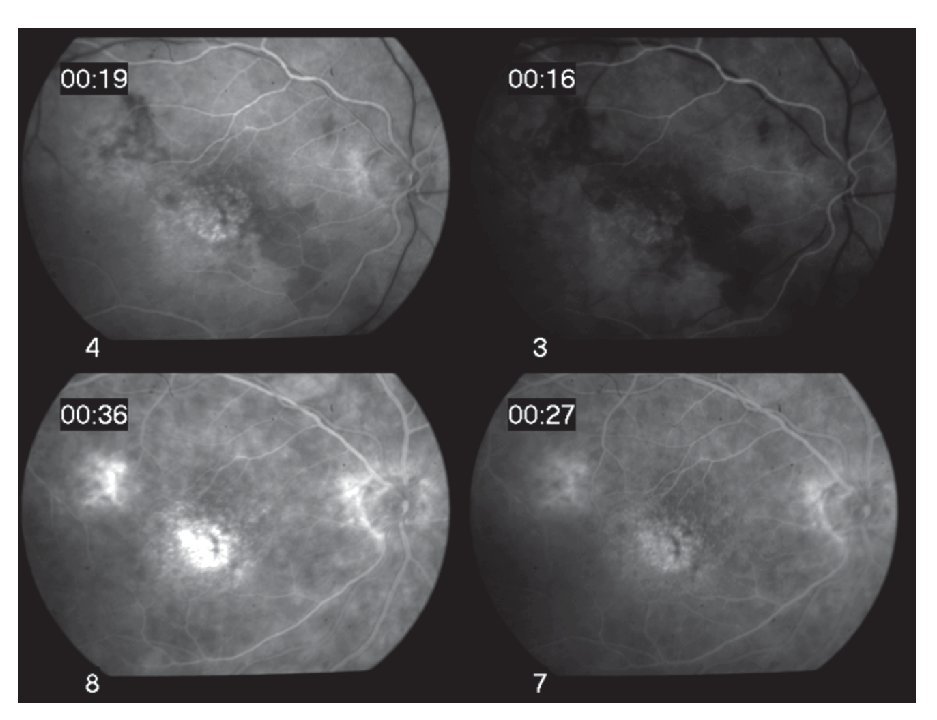
El diagnóstico diferencial incluyó sífilis, coroiditis multifocal y panuveitis, enfermedad de Adamantiades-Behçet, sarcoidosis y coriorretinopatía en perdigonada, entre otras. Estudios de laboratorio, incluyendo biometría hemática, examen general de orina, perfil bioquímico, FTA-Abs, determinación de VIH, anticuerpos antinucleares, fracciones de complemento C3, C4, CH50, ligando de C1q, enzima convertidora de angiotensina, lisozima y tele de tórax, los cuales no mostraron alteración alguna. Sin embargo, la determinación del haplotipo HLA-A29 por el método de microtoxicidad linfocitaria, fue positiva. La angiografía de retina mostró fuga de fluoresceína de la vasculatura retiniana periférica y macular en fases tardías, así como leve hiperfluorescencia en las lesiones coroideas (Figuras 2A y 2B). El ERG mostró una amplitud discretamente reducida, así como un aumento en la latencia de la onda "b".
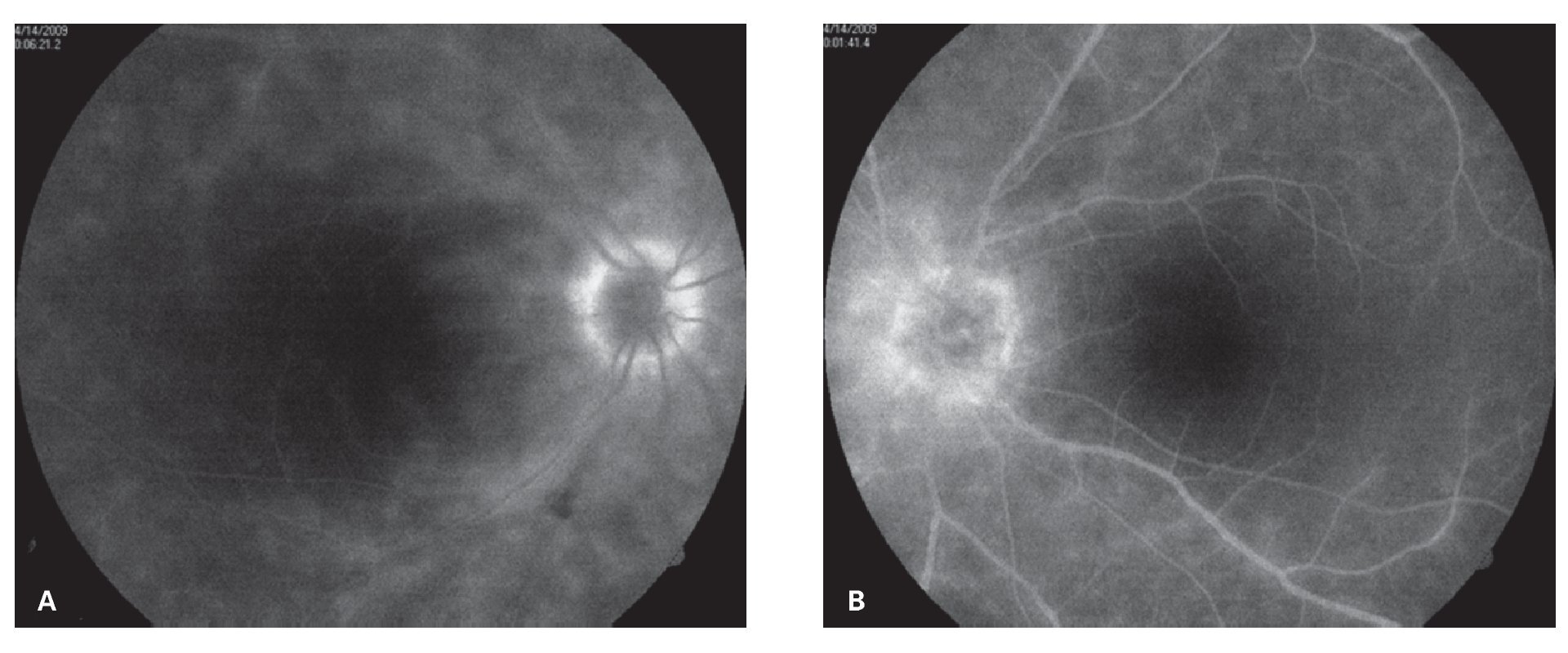
¿ Figura 2. Fase tardía del fluorangiograma (caso 1) del ojo derecho, mostrando tinción y fuga de arteriolas y vénulas en polo posterior, así como del nervio óptico A). Tinción y fuga sectorial de arteriolas y vénulas del ojo izquierdo del mismo paciente B).
Considerando los hallazgos fundoscópicos, fluorangiográficos, el ERG y la presencia del alelo HLA-A29, se llegó al diagnóstico de coriorretinopatía en perdigonada y se inició tratamiento con prednisona (40 mg/día) y azatioprina (100 mg/ día). Debido al antecedente de hipercolesterolemia e hipertensión arterial, se decidió no emplear ciclosporina-A (CsA) como parte del régimen terapéutico. A las tres semanas de iniciado el tratamiento, el paciente manifestó mejoría visual y reducción de las miodesopsias. La visión en el ojo derecho mejoró a 20/25+ y permaneció en el ojo izquierdo en 20/20, la vitreítis se redujo a 1/2+ OU. En esa misma visita, se inició la reducción paulatina de la prednisona hasta llegar a 5 mg/día en un lapso de ocho semanas, manteniendo la azatioprina en 100 mg/día.
En visitas subsecuentes, el paciente mostró una mejoría sintomática considerable, manteniendo una visión no corregida de 20/20 OU, aunque aún notaba un bajo grado de miodesopsias. El paciente se mantuvo en tratamiento con prednisona (2.5 mg/día) y azatioprina (100 mg/día), por 16 semanas. En ese tiempo, las pruebas de función hepática mostraron una discreta elevación de las bilirrubinas (directa = 0.30 mg%, indirecta = 1.58 mg% y total = 1.88 mg%), así como elevación considerable de las transaminasas (glutámico oxaloacética = 580 UI/L, glutámico pirúvica = 680 UI/L y gama glutamiltranspeptidasa = 179 UI/L), lo que obligó a la suspensión inmediata de la azatioprina. Por otra parte, se continuó con la reducción lenta de la prednisona. En consultas subsecuentes, las pruebas de función hepática se normalizaron y la actividad inflamatoria ocular se encontró abolida, manteniéndose la prednisona en 2.5 mg/día por un periodo de seis meses, cuando presenta una nueva reactivación caracterizada por vitreítis de 2+ OD y 1+ OS y periflebitis sectorial, siendo necesario aumentar la prednisona a 30 mg/día y reiniciar la inmunosupresión con azatriopina (50 mg/día). Actualmente el paciente se encuentra asintomático, con una visión mejor corregida de 20/20 OU y no muestra signos de inflamación intraocular.
Caso 2. Masculino de 51 años, quien acude al servicio de inmunología y uveítis por miodesopsias y visión borrosa en ambos ojos, de varios meses de evolución. Al examen oftalmológico inicial, la agudeza visual era de 20/25-2 OU y la biomicroscopía no reveló datos de inflamación, en el segmento anterior. A la fundoscopía indirecta, se observaron múltiples lesiones ovaladas, amarillo cremosas, que parecían radiar del nervio óptico hacia los cuadrantes nasales y que tendían a confluir formando un fondo vitiliginoso (Figuras 3A y 3B). Dichas lesiones se acompañaban de vitreítis difusa 1+, así como papilitis 2+ y periflebitis marcada, que se corroboraron en la fluorangiografía en ambos ojos (Figuras 4A y 4B). Estudios de laboratorio incluyeron, entre otros: anticuerpos antinucleares (ANAs), C3, C4 y CH50, ANCAs, FTA-Abs, PPD, los cuales resultaron negativos o dentro de límites normales. La determinación del haplotipo HLA-A29 resultó positiva, por el método de microtoxicidad de linfocitos. Con estos hallazgos clínicos y la positividad del alelo A-29, se llegó al diagnóstico de coriorretinopatía en perdigonada. Se inició tratamiento empleando prednisona (60 mg/ día), con reducción paulatina hasta 10 mg/día, en un lapso de seis semanas y azatioprina (100 mg/ día). En un periodo de 10 semanas bajo este tratamiento, el paciente refirió mejoría significativa de la visión y miodesopsias, encontrándose una agudeza visual de 20/20-2 OU, no había inflamación en el segmento anterior y la vitreítis se redujo a 1/2+ OU. Aunque las lesiones coriorretinianas permanecían igual, los márgenes de ambos nervios ópticos se encontraron bien definidos y sin edema, observándose un ligero envainamiento (fibrosis) perivascular OU. En los siguientes seis meses, el paciente permaneció asintomático, manteniendo una agudeza visual de 20/20 en ambos ojos y sin datos de inflamación en el segmento posterior. El tratamiento se mantuvo con prednisona (2.5 mg/día) y azatioprina (100 mg/día) por un total de 10 meses, antes de su suspensión. El paciente no ha vuelto a consulta, desde la suspensión del tratamiento.
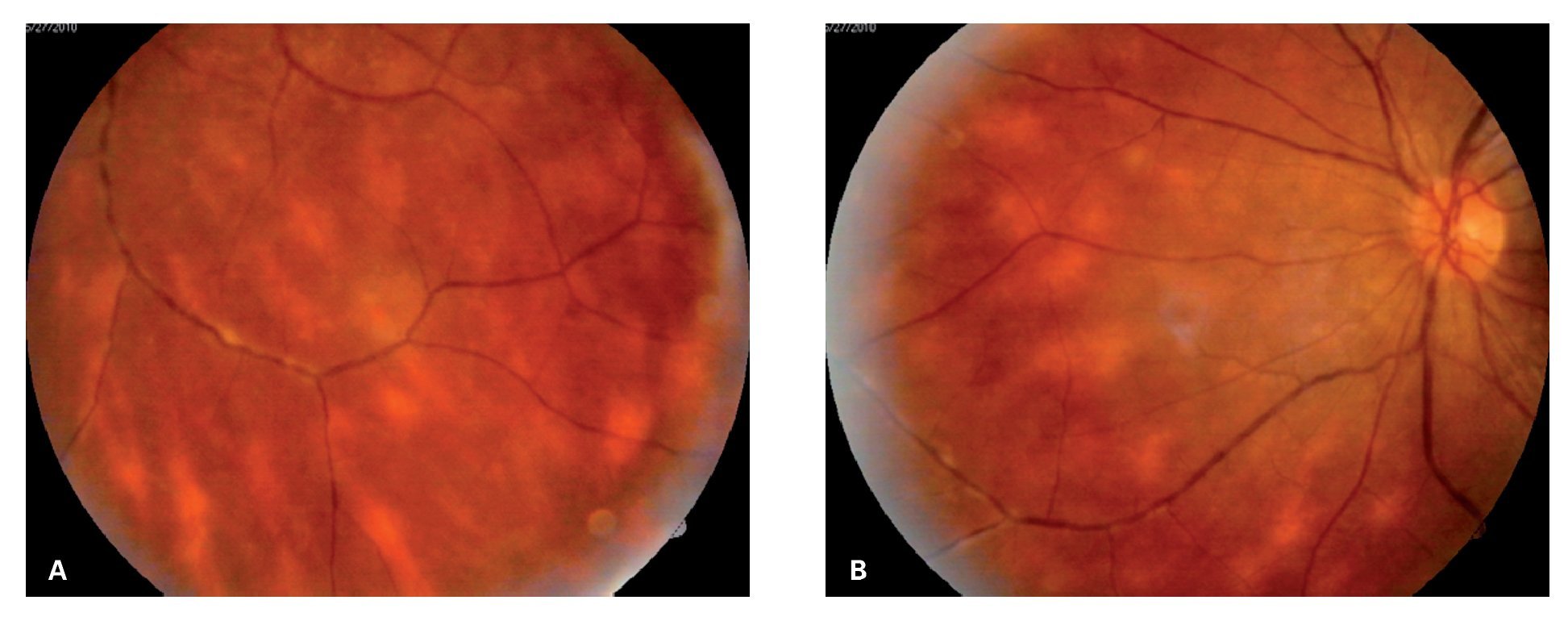
¿ Figura 3. Múltiples lesiones amarillo-cremosas de distribución radial al nervio óptico con dirección hacia los cuadrantes nasales, características de cororretinopatía en perdigonada en ojo derecho (Caso 2) A). Imagen en espejo de las mismas lesiones en el ojo izquierdo del mismo paciente. La falta de nitidez de las imágenes se debe a la vitreítis B).
¿ Figura 4. Fase tardía del fluorangiograma (caso 2), mostrando tinción y fuga vascular generalizada, así como del nervio óptico del ojo derecho A). Papilitis izquierda, evidenciada por la extensa fuga de fluoresceína en la fase tardía extrema B).
Caso 3. Femenina de 64 años de edad que acude a consulta, con antecedentes de panuveítis bilateral crónica de cinco años de evolución. La paciente había sido tratada previamente con dosis altas de esteroides tópicos y sistémicos, presentando síndrome de Cushing, sobrepeso e insomnio. Además, fue operada de extracción de catarata secundaria a esteroides en su ojo derecho. La paciente acudió a nuestra clínica para una segunda opinión por visión borrosa y miodesopsias en ambos ojos, así como por los efectos secundarios al tratamiento previo. A su examen oftalmológico la agudeza visual mejor corregida era de 20/150 OD y 20/40 OS. La visión de colores mostró discromatopsia al verde OU. El examen biomicroscópico reveló trazas de células OD y 1/2+ OS, en cámara anterior. Se observó lente intraocular de cámara posterior, en la bolsa con buen centrado OD. A la fundoscopía, el ojo derecho presentó 3+ vitreítis, mientras que en el ojo izquierdo, el vítreo estaba claro. En la retina periférica media de ambos ojos, se observaron múltiples lesiones de forma oval, de color amarillo pálido y de distribución nasal superior e inferior, inmediatamente por fuera de las arcadas vasculares, las cuales eran coalescentes produciendo un fondo "vitiliginoso" OU (Figuras 5A y 5B). La determinación serológica del haplotipo HLA-A29, por reacción en cadena de polimerasas (PCR) resultó positiva.
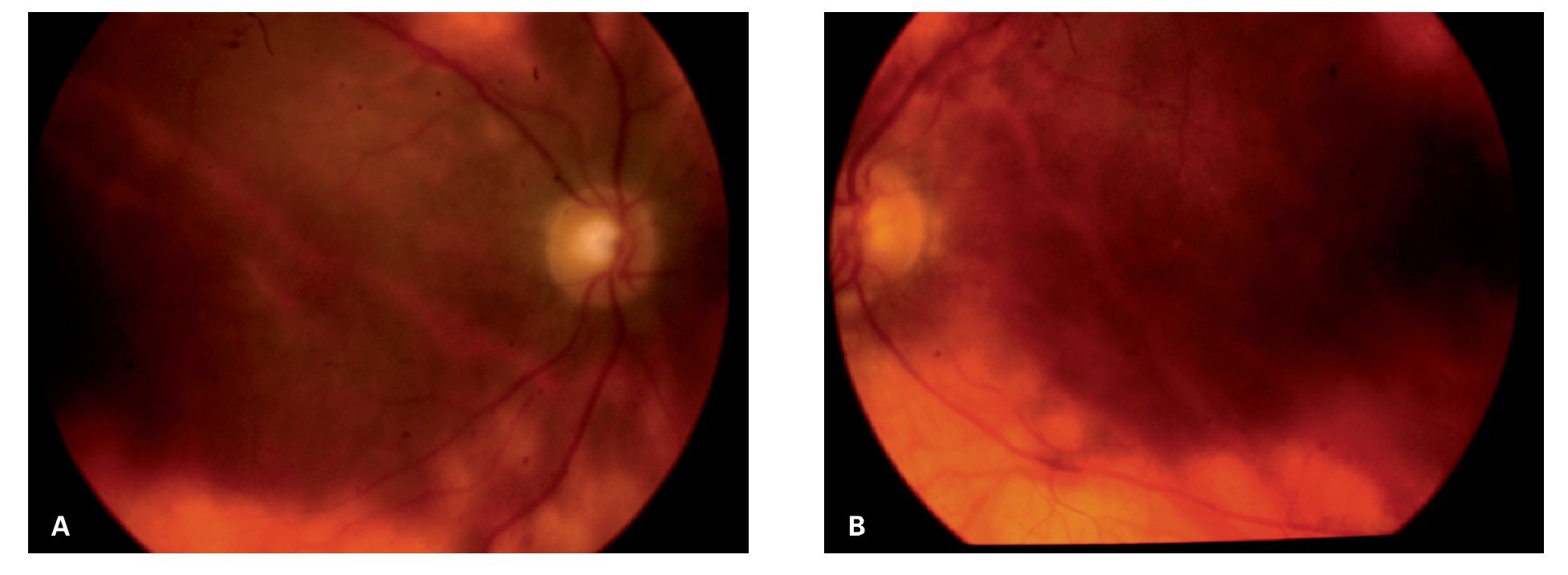
¿ Figura 5. Fondo de ojo derecho (caso 3) con vitreítis 2+ y lesiones típicas de coriorretinopatía en perdigonada de distribución nasal al nervio óptico, confluyendo y dando una apariencia vitiliginosa A). Afectación del ojo izquierdo que muestra con mayor claridad dicha apariencia de la enfermedad B).
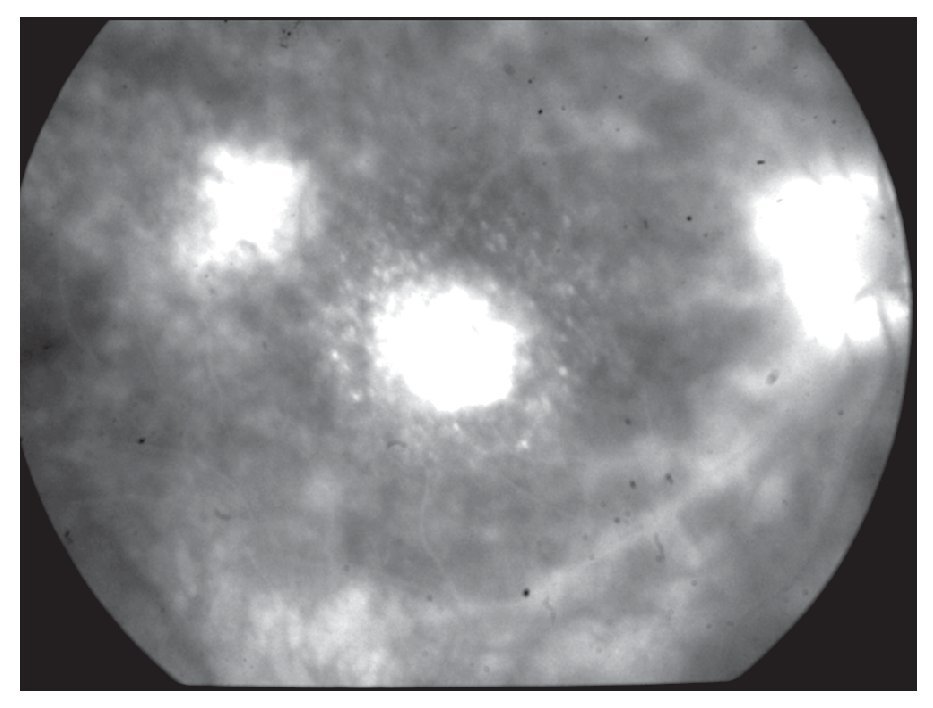
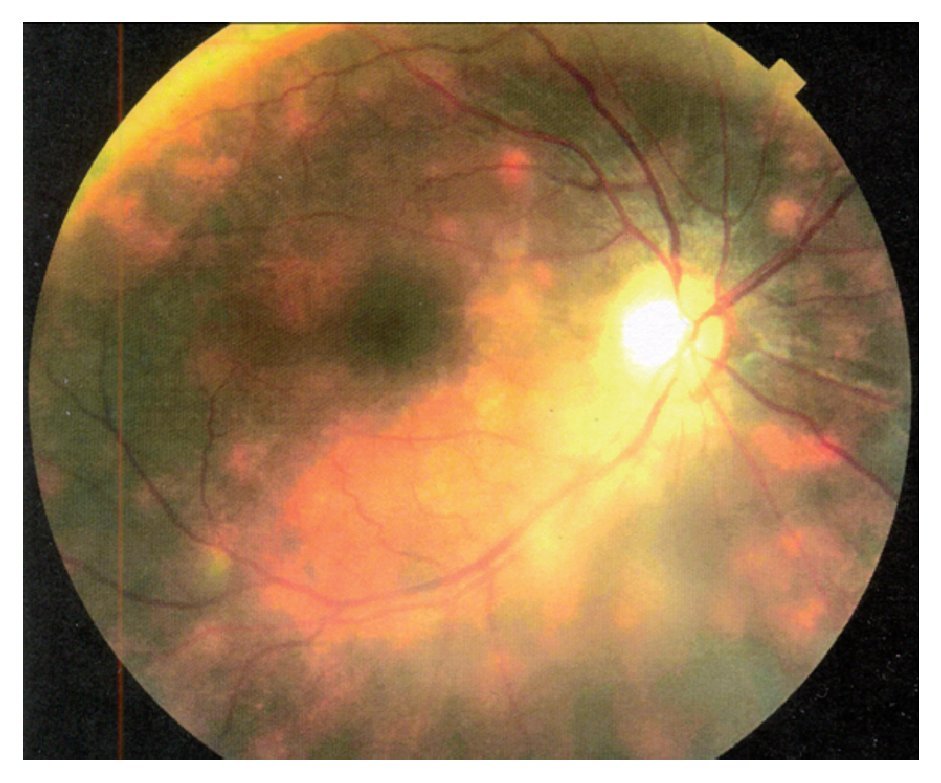
Dados los antecedentes de uveítis crónica bilateral con exacerbaciones y remisiones, así como los hallazgos oftalmológicos y la presencia del alelo A29, se llegó al diagnóstico de coriorretinopatía en perdigonada. Se inició tratamiento con CsA (325 mg/día, vía oral) y azatioprina (100 mg/ día), así como destete paulatino de la prednisona. Un mes después de iniciado el tratamiento, la paciente refirió mejoría sintomática, aunque la agudeza visual no mostró cambio alguno. Al examen oftalmológico no se encontró inflamación en cámara anterior OU, además de que fue evidente la disminución de la vitreítis a 1+ OD. Tres meses después de iniciado este régimen terapéutico, la paciente regresa asintomática con una agudeza visual de 20/100 OD y 20/40 OS. En ese momento, se encontraba con CsA (300 mg/día) y azatioprina (100 mg/día). La paciente refirió temblor fino ocasional en ambas manos e hirsutismo. Nueve meses después, acude a consulta con disminución de la visión en ojo derecho a 20/800, debido al desarrollo de catarata subcapsular posterior 3+, sin embargo mostró una mejoría a 20/30 en ojo izquierdo. A la fundoscopía del ojo derecho se observaron alteraciones maculares sugestivas de neovascularización coroidea, la cual se confirma por medio de un fluorangiograma (Figura 6). Se decide inyectar 40 mg de acetónido de triamcinolona por vía transeptal, en dicho ojo y mantener el mismo régimen terapéutico. Cabe destacar, que en ese tiempo aún no había disponibilidad de terapia antiangiogénica. La paciente regresa un año después, con una cicatriz disciforme en el área macular del ojo derecho, vasculitis retiniana y papilitis activa OS (Figura 7). En ese momento no se encontraba en terapia inmunosupresora. La paciente cambió de país de residencia, por lo cual se refirió a un centro de alta especialidad en su lugar de destino.
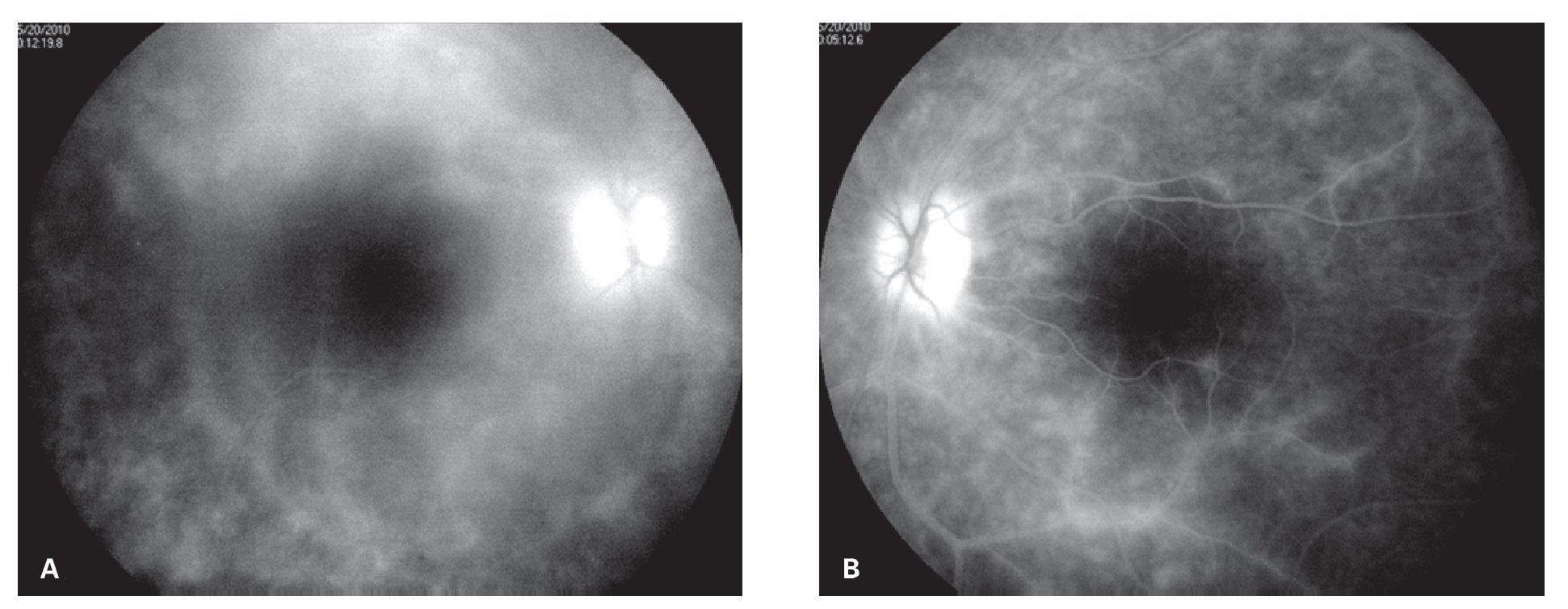
¿ Figura 6. Angiografía fluorescente de retina (Caso 3), mostrando neovascularización coroidea macular del ojo derecho. Se aprecia además defecto de ventana por atrofia del epitelio pigmentario de retina en el área temporal, así como discreta fuga de colorante en área peripapilar.
¿ Figura 7. Fase tardía de fluorangiografía de retina (Caso 3) mostrando tinción y fuga difusa perivascular, así como del nervio óptico del ojo derecho. Se observan además defectos de ventana en áreas de atrofia y cicatrización en la fóvea por membrana neovascular previa y temporal a la misma por atrofia del epitelio pigmentario de retina.
Caso 4. Femenina de 37 años de edad, que acude a consulta con historia de panuveítis bilateral idiopática, de seis años de evolución. La paciente estaba siendo atendida previamente en otro centro oftalmológico, presentando un curso clínico crónico de uveítis caracterizado por exacerbaciones y remisiones múltiples. Había sido tratada previamente con esteroides tópicos por tiempo prolongado, múltiples (>8) inyecciones sub-tenonianas de acetónido de triamcinolona (40 mg/dósis) en cada ojo, así como esteroides sistémicos con pobre respuesta terapéutica y que provocaron sobrepeso excesivo, síndrome de Cushing y diabetes mellitus secundaria. Además, la paciente presentó cataratas secundarias en ambos ojos, por lo cual fue operada de extracción extra-capsular, sin implantación de lentes intraoculares, con pobre resultado visual. También presentó glaucoma secundario por bloqueo pupilar OS, el cual se resolvió al extraer la catarata.
En su primera visita a nuestro servicio, la paciente refería visión borrosa progresiva, fotofobia y cefalea. El examen oftalmológico reveló una agudeza visual corregida, con lentes de contacto blandos de 20/400 OD y movimiento de manos OS. A la biomicroscopía se encontró celularidad de 1/2+ OD y 2+OS y fenómeno de Tyndall 4+ OU. Las pupilas eran irregulares, con sinequias anteriores periféricas múltiples OU. Además presentaba afaquia en ambos ojos. A la fondoscopía se encontró vitreítis de 2+ OD y 3+ OS, así como múltiples lesiones coriorretinianas ovaladas (200-350 m de diámetro), de coloración cremaamarillentas y distribución peripapilar, con predominancia nasal en el ojo derecho. Además, se observó fibrosis perivascular segmentaria leve en el mismo ojo. En ojo izquierdo, la vitreítis (3+) no permitía observar detalles del fondo del ojo. La presión intraocular era de 12 mmHg OD y 8 mmHg OS. Debido a la historia previa y cuadro clínico, se consideraron como diagnósticos posibles: el síndrome de Vogt-Koyanagi-Harada (VKH), la coriorretinopatía en perdigonada, la coroiditis multifocal y panuevítis, la sífilis, entre otros. Una biometría hemática, perfil bioquímico y examen general de orina resultaron normales. El VDRL y FTA-Abs fueron negativos. Un ultrasonido A-B del ojo izquierdo, mostró imágenes hiperecóicas homogéneas consistentes con vitreítis, la retina se encontró aplicada sin desprendimiento exudativo, ni engrosamiento coroideo. A la paciente se le solicitó angiografía fluorescente de retina y determinación del haplotipo HLA-A29, sin embargo no los realizó por dificultades económicas.
Considerando que la paciente no cumplía con los criterios diagnósticos revisados por el Comité de Estudio de Nomenclatura del síndrome de VKH37, así como los hallazgos ultrasónicos del ojo izquierdo y la pobre respuesta a esteroides, este diagnóstico era improbable. Por otro lado, las lesiones coriorretinianas no tenían características morfológicas típicas "en sacabocado", hipertrofia del epitelio pigmentario, tamaño variable y distribución más periférica con predominio por cuadrantes inferiores, que hicieran pensar en coroiditis multifocal y panuveítis. El curso clínico crónico con exacerbaciones y remisiones, las lesiones blanco-amarillentas de distribución peripapilar con predominio nasal, el fondo de apariencia vitiliginosa y los cambios vasculares de la retina, apuntaron a una coriorretinopatía en perdigonada.
La paciente no tenía posibilidades económicas de recibir tratamiento con CsA, por lo cual se inició tratamiento con azatioprina (150 mg/día) como monoterapia, atropina al 1% cada noche, acetato de prednisolona al 1% cada ocho horas, obteniéndose una mejoría de su cuadro clínico en los siguientes 10 meses. Para entonces, la agudeza visual mejor corregida del ojo derecho era 20/200 y en ojo izquierdo 20/800, los signos de inflamación tanto del segmento posterior como anterior, cedieron por completo. A cuatro años de su primer visita en nuestro servicio, la agudeza visual mejor corregida es de 20/200 OD y 20/300 OS, no presentando signos de inflamación activa.
Caso 5. Masculino de 58 años de edad, quien fue referido a nuestro servicio con historia de vasculitis y coriorretinitis bilateral crónica, de etiología desconocida y difícil manejo. El paciente había sido tratado previamente con gotas de esteroides en ambos ojos, prednisona (40 mg/día) y CsA (300 mg/día, vía oral) por dos años. Además, había recibido fotocoagulación periférica de retina con laser argón en ambos ojos, por una vasculitis oclusiva retiniana. Como antecedentes de importancia, presentaba hipertensión arterial sistémica y osteoma de células gigantes.
Al examen oftalmológico inicial, presentó una agudeza visual mejor corregida de 20/60 OD y 20/100 OS. La biomicroscopía reveló una inyección ciliar de 2+ OU y en la cámara anterior del ojo derecho, se observaron trazas de células inflamatorias y sinequias posteriores del iris. En el ojo izquierdo, había 1/2+ de células. En ambos ojos se observó la formación de cataratas subcapsulares posteriores (1+). La presión intraocular se encontró en 15 mmHg OU. En el fondo de ojo, se apreciaron múltiples lesiones coriorretinianas redondeadas de coloración amarillo-cremoso, con predominio por los cuadrantes nasales, así como huellas dispersas de fotocoagulación con laser argón en ambos ojos.
En ese momento, el paciente no tenía un diagnóstico definitivo, considerándose a la coroidopatía multifocal, panuveítis y coriorretinopatía en perdigonada, como causas más probables. Un VDRL y FTA-Abs resultaron negativos, pero el haplotipo HLA-A29 dio positivo. Se continuó tratamiento con CsA (100 mg/día), agregándose azatioprina (150 mg/día). Además, se inició destete de esteroides tópicos empleando acetato de fluorometolona al 1%, se continuó tratamiento con betaxolol 0.5% cada 12 horas, y brimonidina 0.1% cada 12 horas, en ambos ojos.
Tres semanas después, el paciente regresa a consulta refiriendo visión borrosa en ambos ojos. En ese momento, la agudeza visual corregida era de 20/50 OD y 20/150 OS. A la fundoscopía del ojo izquierdo, se observan microaneurismas en el sector nasal inferior y edema macular quístico. Una angiografía fluorescente mostró hipoperfusión capilar moderada e hipoxia discreta, en áreas dispersas de la retina de ambos ojos, así como tinción y fuga de vasos sanguíneos y acumulo del material de contraste, compatible con edema macular quístico en ojo izquierdo. Dado el curso clínico crónico de su enfermedad y las manifestaciones clínicas caracterizadas por uveítis bilateral crónica y recurrente, coriorretinitis multifocal, vasculitis oclusiva con edema macular en ojo izquierdo, así como la presencia del haplotipo HLA-A29, se llegó al diagnóstico de coriorretinopatía en perdigonada. Se continuó tratamiento sistémico con CsA (100 mg/día) y azatioprina (150 mg/día), manteniendo el tratamiento tópico igual. Dos y medio meses después, regresa con mejoría sintomática significativa, presentando una agudeza visual de 20/50 OD y 20/100 OS. No se encontró inflamación en segmento anterior, ni en cavidad vítrea, su presión intraocular era de 17 mmHg OU. Cuatro meses después, acude a valoración por disminución de la visión en ojo izquierdo (20/100-2), así como deslumbramiento secundario a catarata en el mismo ojo, por lo que se decide realizar facoemulsificación de catarata e implantación de lente intraocular. La cirugía resulta exitosa, lográndose una mejoría visual de 20/30 OS. En los siguientes ocho meses, el paciente se mantuvo en tratamiento con CsA (100 mg cada tercer día) y azatioprina (50 mg cada tercer día) alternándose, así como brinzolamida cada 12 horas y betaxolol 0.5% cada 12 horas OU. Los ojos permanecieron sin inflamación por todo ese periodo, sin embargo desarrolló catarata subcapsular posterior en el ojo derecho, resultando en una visión de 20/200. En su última visita, el paciente se encontraba fuera de la CsA y justo, había suspendido la azatioprina por estomatitis herpética oral recurrente. Tres meses después de esta última visita, el paciente fallece a consecuencia de complicaciones del osteoma de células gigantes que venía padeciendo previamente.
Caso 6. Femenina de 43 años de edad con antecedentes de hipertensión arterial sistémica y diabetes mellitus secundaria, quien inicia hace cuatro años con visión borrosa y miodesopsias de ambos ojos, por lo que acudió a un centro oftalmológico en donde fue diagnosticada como uveítis crónica bilateral. La paciente fue tratada con prednisona (50 mg/día) por periodos largos de tiempo (mayor a seis meses), así como múltiples inyecciones perioculares de esteroides de depósito y esteroides tópicos, por tiempo indefinido sin mostrar resultados satisfactorios. Al momento de la consulta en nuestro servicio, presentaba una agudeza visual de 20/400 OD y movimiento de manos OS. A la biomicroscopía se encontró celularidad de 4+ en cámara anterior OU, así como catarata 3+ OS, que no permitía observar detalles del fondo del ojo. La fondoscopía reveló una vitreítis 2+ OD y 4+ OS, lesiones coriorretininanas redondeadas de coloración amarillenta alrededor del nervio óptico y un fondo vitiliginoso, en ojo derecho (Figura 8). La presión intraocular se encontraba en 12 mmHg OD y 18 mmHg OS. Debido al curso clínico crónico-recurrente, las características de las lesiones coriorretinianas observadas en el ojo derecho y la pobre respuesta terapéutica a los esteroides, el diagnóstico diferencial incluyó a la coriorretinopatía en perdigonada, entre otras. Se realizaron estudios auxiliares de diagnóstico consistentes en: PPD, BAAR en orina, VDRL, FTA-Abs, biometría hemática, perfil bioquímico. Sólo se encontró hiperglucemia e hipercolesterolemia, siendo el resto de los estudios negativos o dentro de rangos normales. Una fluorangiografía de retina mostró tinción y fuga tardía del área peripapilar, consistente con papilitis y de vasos retinianos y edema macular quístico OD. Un ultrasonido A-B del ojo izquierdo, reveló desprendimiento de vítreo posterior y vitreítis densa. Se realizó determinación del haplotipo HLA-A29, el cual fue positivo por reacción en cadena de polimerasas (PCR). Dadas las características fundoscopías del ojo derecho y la presencia del alelo A29, se diagnosticó coriorretinopatía en perdigonada en etapa avanzada. Se inició tratamiento a base de azatioprina (100 mg/día), así como ciclofosfamida (750 mg/pulso-IV por seis). Se decidió no emplear CsA, debido a los antecedentes de hipercolesterolemia e hipertensión arterial sistémica. En las siguientes tres semanas, la paciente no sintió mejoría significativa, a pesar de que la visión del ojo izquierdo mejoró a 20/800 y la vitreítis del ojo derecho disminuyó levemente. Se incrementó la azatioprina a 150 mg/ día y a la octava semana de tratamiento, la visión mejoró a 20/200 OD y 20/400 OS. Actualmente, la paciente se encuentra sin inflamación intraocular en ambos ojos y en espera de facoemulsificación de catarata e implante de lente intraocular OS.
¿ Figura 8. Apariencia vitiliginosa del fondo de ojo derecho (Caso 6), mostrando múltiples lesiones coriorretinianas de coloración amarillo-cremoso y distribución radial al nervio óptico, características de retinopatía en perdigonada.
¿ Discusión
La coriorretinopatía en perdigonada (birdshot) es una forma de uveítis posterior crónica, cuyo origen se presume "autoinmune", sustentado en la presencia de hiperreactividad sérica de pacientes afectados contra el antígeno soluble retiniano "S", en la presencia de altas concentraciones de IL-17, IL-2, IL-6 y TNF-alfa, en el humor acuoso de estos pacientes. Así como en la probable susceptibilidad inmuno-genética fuertemente ligada al haplotipo HLA-A29.14,15,29
Se trata de una enfermedad rara, representando solamente del 0.6% al 1.5% de todos los casos de uveítis, referidos a centros de alta especialidad.7 Además, la gran mayoría de los casos reportados en la literatura médica son caucásicos, descendientes del norte de Europa, excepto por un paciente de origen hispano y otro de raza negra, documentados en dichas series.10 En México, sólo se ha reportado un caso aislado de la enfermedad en una paciente femenina de 45 años de edad, quien presentó lesiones típicas a la fondoscopía.38 De esta manera, antes del presente reporte, sólo tenemos conocimiento de dos casos aislados de pacientes de origen hispano, presentados en la literatura oftalmológica.10,38 Los seis pacientes presentados en este reporte, son todos de origen hispano, oriundos del noreste de México (Tabla 1).
Mientras reportes previos han mostrado un ligero predominio del género femenino en esta enfermedad, con un rango que va del 51.7% al 72.7% (promedio 54.1%),10 en el presente encontramos igualdad de género. Típicamente, la coriorretinopatía en perdigonada ocurre en pacientes de edad adulta media. En el presente reporte, la edad promedio fue un poco más baja (47.2 años), comparado con 53 años de todos los casos previamente reportados, en la literatura médica.10
Los síntomas iniciales de la coriorretinopatía en perdigonada incluyen, visión borrosa secundaria a vitreítis y/o a edema macular y miodesopsias, ocasionadas por la propia vitreítis. Los pacientes también se pueden quejar de nictalopía, fotopsias, deslumbramiento y discromatopsia.3,4 Ambos ojos están afectados, aunque el compromiso puede ser asimétrico. Es importante recalcar, que algunos pacientes pueden presentar molestias mínimas o simplemente permanecer asintomáticos por periodos largos de tiempo.6 En nuestra población, los síntomas predominantes fueron visión borrosa y miodesopsias presentes en todos los casos, además una paciente presentó fotofobia y dos de ellos (Casos 1 y 3), discromatopsia caracterizada por desaturación al color rojo y verde, respectivamente.
El diagnóstico de la enfermedad está basado en las manifestaciones clínicas, así como también en los hallazgos oftalmoscópicos.8 En la Tabla 2 se muestran los hallazgos encontrados en esta serie de casos, entre los que predominan la vitreítis, la vasculitis retiniana y las típicas lesiones coroideas, que representan la característica diagnóstica más importante. Además en esta serie, seis de los 12 ojos (50%) estudiados, presentaron al menos un episodio de inflamación del nervio óptico.
El caso previamente reportado en la literatura mexicana, presentó características clínicas típicas de coriorretinopatía en perdigonada, y aunque la evaluación diagnóstica incluyó a la fluorangiografía y el ERG, cabe destacar que no se mencionó la determinación del haplotipo HLA-A29.38 En la presente serie, en los cinco pacientes que se realizaron la determinación del haplotipo HLA-A29, éste resultó positivo, confirmando el diagnóstico y mostrando la clara asociación que existe, entre este alelo y la enfermedad. Es importante considerar, que este antígeno sólo se encuentra en el 7% de la población general, mientras que está presente en el 98% de los pacientes con coriorretinopatia en perdigonada.15,24
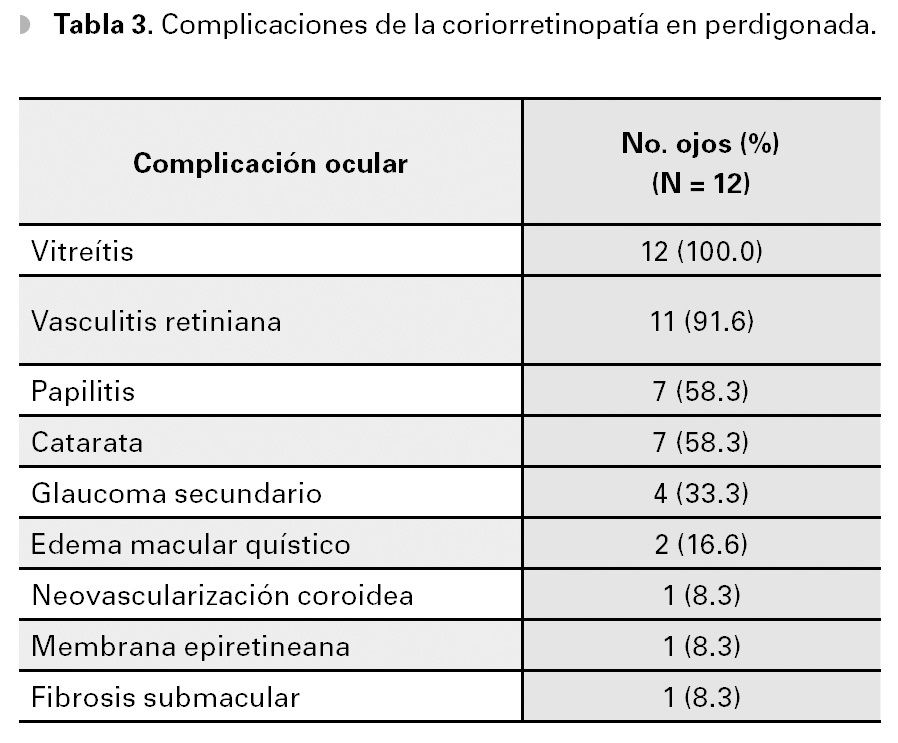
Las complicaciones de esta enfermedad incluyen a la vitreítis, la vasculitis retiniana, la papilitis, el edema macular quístico, la neovascularización coroidea macular (CNV) y la formación de membrana epiretiniana, entre otras.3,8 En el presente estudio, todos los ojos presentaron algún grado de vitreítis (Tabla 2). Además, la angiografía demostró tinción y fuga de vasos retinianos en 11 (91.6%) ojos, así como papilitis en siete (58.3%) ojos. Edema macular quístico se observó en dos (16.6%) ojos, CNV y la formación de membrana epirretiniana, en un ojo cada una (Tabla 3). Además, siete (58.3 %) ojos de cuatro pacientes presentaron formación de catarata secundaria y cuatro (33.3%) ojos de dos pacientes, glaucoma secundario (Tabla 3).
En lo que respecta a la agudeza visual, se encontró una gran diferencia entre aquellos pacientes en quienes se llevó a cabo un diagnóstico y tratamiento oportuno, comparado con aquellos en quienes el diagnóstico se retrasó por meses o años, y el manejo no fue el adecuado desde el inicio de la enfermedad. En esta serie de casos, sólo dos pacientes acudieron a consulta en etapas tempranas de la enfermedad (Casos 1 y 2), mientras que los otros cuatro pacientes (Casos 3 a 6), fueron vistos en etapas avanzadas del curso clínico. La agudeza visual inicial y final de los dos primeros casos, fue en rango de 20/20 a 20/30, el cual contrastó con el resto de los pacientes (Casos 3 a 6), en los cuales la agudeza visual varió entre 20/40, hasta movimiento de manos. Este hallazgo confirma el hecho de que el pronóstico visual en esta forma de uveítis, al igual que en la mayoría de las causas de inflamación intraocular crónica, es peor cuando existe un retraso en el diagnóstico etiológico y el tratamiento adecuado.
Por otra parte, existe evidencia de que la disfunción retiniana crónica progresiva que presentan los pacientes con coriorretinopatía en perdigonada puede ocurrir aún en presencia de una agudeza visual estable, ausencia de edema macular o vitreítis, llevando al paciente a una pérdida visual al mediano o largo plazo.21 Razón por la cual, es recomendable establecer revisiones clínicas periódicas, acompañadas de estudios campimétricos automatizados y electrofisiológicos, durante su seguimiento.21,32
En un principio, cuando se conocía poco sobre esta enfermedad, el tratamiento estaba dirigido a tratar las exacerbaciones inflamatorias y el edema macular quístico. Sin embargo, con la evidencia reciente de que puede ocurrir disfunción retiniana crónica en presencia de una agudeza visual estable, la estrategia de tratamiento ha cambiado a favor del empleo prolongado de inmunosupresores.21,32,36,39
Se ha descrito el tratamiento con esteroides orales a dosis de 40 a 60 mg/día, disminuyendo de forma paulatina al presentar mejoría visual y agregando, en caso necesario, inyecciones sub-tenonianas de depósito.10,23,40 Una tercera parte de los ojos tratados de esta manera perdieron dos o más líneas de visión, mientras que un poco más de la mitad de los ojos, tuvieron cambio en la agudeza visual de apenas una o menos líneas de Snellen, en un tiempo de seguimiento promedio de 68.4 meses.10
En la presente serie, tres de los pacientes (Casos 3, 4 y 6), habían sido tratadas previamente con dosis altas de esteroides sistémicos y perioculares, presentando un pobre resultado terapéutico y efectos adversos caracterizados por sobrepeso, síndrome de Cushing, diabetes mellitus, insomnio, hirsutismo, dermatitis acneiforme, entre otros, al momento de su primer visita a nuestro servicio. Además, estos pacientes presentaron el peor resultado visual de los seis casos reportados, con un rango visual final de 20/40 a 20/800.
Debido al curso clínico crónico de la coriorretinopatía en perdigonada, los pacientes requieren terapia prolongada, por lo que el uso de esteroides no es la mejor alternativa, debido a los efectos adversos que produce a dosis altas y a la pobre respuesta terapéutica, antes mencionada.10,23 Entonces, los esteroides sólo son útiles como coadyuvante en las etapas de exacerbación de la inflamación, reduciendo su administración en cuatro a seis semanas, a dosis bajas (5-10 mg/día), lo que hace necesaria la administración de terapia inmunosupresora clásica.23,34
La CsA surgió como alternativa terapéutica para la coriorretinopatía en perdigonada, debido a su mecanismo de acción inhibitorio sobre linfocitos-T mediante el bloqueo de interleucina-2 (IL-2).34,39 Este medicamento ha mostrado prevenir la uveítis experimental autoinmune inducida por antígeno-S.13 Le Hoang y colaboradores trataron 21 pacientes con coriorretinopatía en perdigonada, administrando CsA a una dosis de 5-10 mg/kg/ día, encontrando una reducción significativa en la inflamación intraocular y en algunos casos, mejoría en la campimetría y el ERG.39 Por otra parte, Vitale y colaboradores describieron el tratamiento de 13 pacientes con coriorretinopatía en perdigonada, empleando CsA a dosis baja (2.5-5 mg/kg/ día).34 Cinco de estos pacientes requirieron la adición de azatioprina (1.5-2 mg/kg/día), para lograr control inflamatorio. Estos autores reportaron mejoría o estabilidad visual en 20 ojos (83.3%) tratados con CsA, comparado con cinco ojos (45.5%) que no recibieron el medicamento. Ningún paciente desarrolló nefrotoxicidad, aunque dos de ellos presentaron hipertensión arterial.34 En la presente serie de casos, la CsA fue administrada a dosis baja (2.5-5 mg/kg/día) en dos pacientes (Casos 3 y 5) con éxito, permitiendo reducir y eliminar el uso de esteroides sistémicos. En otros dos pacientes (Casos 1 y 6), la CsA no pudo ser empleada debido a que estos pacientes padecían hipertensión arterial e hipercolesterolemia. Finalmente, los dos pacientes restantes (Casos 2 y 4) no recibieron tratamiento con CsA, debido a dificultades económicas. Se puede concluir que el tratamiento de la coriorretinopatía en perdigonada con CsA en combinación con azatioprina cuando sea necesario, y limitando el uso de prednisona a dosis bajas, es significativamente mejor que con esteroides como monoterapia.
Otros agentes inmunosupresores, que han sido empleados como monoterapia o en combinación con otros antiinflamatorios para la coriorretinopatía en perdigonada son la azatioprina,8,13,23,34,40,41 la ciclofosfamida,8,13,42 el clorambucilo,13,42 y el micofenolato mofetilo.23 En esta serie de casos, todos los pacientes recibieron tratamiento con azatioprina en combinación con dosis bajas de prednisona (5-10mg/día) y/o de CsA (2.5-5 mg/kg/día), además de que una paciente (Caso 6), requirió de ciclofosfamida intravenosa (1 g/pulso-IV cada seis semanas por seis dosis), debido a la gravedad y lo avanzado del proceso inflamatorio, en que se encontraba al momento de su primer visita en nuestro servicio. Lo que se puede concluir respecto al uso de agentes inmunosupresores en esta enfermedad, es que el número de pacientes tratados es muy pequeño y por tanto, al igual que lo reportado en la literatura respecto a la respuesta terapéutica, existe una gran limitación para realizar un análisis contundente y sacar conclusiones claras de su eficacia en esta patología.
Otra alternativa terapéutica para la coriorretinopatía en perdigonada, ha sido propuesta también por LeHoang y colaboradores,43 quienes describieron el tratamiento de 18 pacientes con inmunoglobulina intravenosa (IVIG), en un tiempo promedio de seguimiento de 39 meses (rango entre 12 a 53 meses). En 14 de 26 ojos con visión de 20/30 o peor, la agudeza visual mejoró en dos o más líneas de Snellen. De 26 ojos con campimetrías anormales, 20 (76.9%) mostraron una mejoría en el campo visual posterior al tratamiento con IVIG. El edema macular mejoró en 17 de 23 ojos (73.9%) a seis meses.43 Los efectos adversos durante el tratamiento con IVIG incluyeron: fiebre, taquicardia e hipertensión arterial.43,44
Respecto al edema macular quístico, éste causa frecuentemente pérdida visual en pacientes con coriorretinopatía en perdigonada.3,6,9 Thorne y colaboradores22 sugieren que la terapia inmunosupresora, puede reducir el riesgo de la aparición de edema macular quístico. También se han reportado buenos resultados con triamcinolona intravítrea.45 Además, un dispositivo intravítreo de liberación prolongada de acetónido de fluocinolona, también ha sido empleado en el tratamiento de la coriorretinopatía en perdigonada.46 Sin embargo, a pesar de que se reportó un éxito absoluto en el control del proceso inflamatorio en 22 ojos, todos los pacientes (100%) que continuaron un seguimiento de por lo menos tres años, desarrollaron cataratas secundarias, y todos (100%) desarrollaron glaucoma secundario para los 12 meses, de instalado el dispositivo. Por lo cual, el 87% de ellos requirieron tratamiento médico antiglaucomatoso y 28% cirugía filtrante o valvular.46
¿ Conclusiones
El presente reporte muestra la casuística más extensa de pacientes hispanos con coriorretinopatía en perdigonada, descrita en la literatura médica hasta el momento. La previa escasez de reportes de la enfermedad, puede deberse a su relativa rareza, la cual es constatada en las series publicadas en la literatura mundial.
La demografía y forma de presentación de la enfermedad, no es distinta de lo reportado en pacientes caucásicos, siendo la edad media de inicio de la enfermedad en esta serie ligeramente menor (47.2 años), que la reportada por otros autores. Por otra parte, la visión borrosa y las miodesopsias fueron los síntomas de inicio más frecuentes, al igual que describen otros autores. En cuanto a las manifestaciones clínicas de la enfermedad, éstas se presentaron en concordancia con lo descrito en publicaciones clásicas como la de Ryan y Maumenee.
Sin embargo, en la presente serie encontramos un espectro clínico más extenso, observando dos pacientes desde el inicio de la enfermedad y otros cuatro, que presentaron un mayor número de complicaciones graves y pérdida visual significativa debido a cataratas, edema macular quístico, papilitis, neovascularización coroidea, entre otros. Esto refleja las consecuencias del retraso en el diagnóstico y tratamiento de la enfermedad. Cuando el diagnóstico y tratamiento es oportuno y adecuado (Casos 1 y 2), el resultado visual y la ausencia de complicaciones es evidente.
Esta enfermedad es crónica y recurrente, razón por la cual se requiere de tratamiento a largo plazo, primordialmente con inmunoterapia clásica para el control adecuado y la disminución de complicaciones potenciales.
Finalmente, el monitoreo de la progresión de la enfermedad debe basarse no solo en la agudeza visual, sino en la progresión observada en la campimetría, la electrofisiología y el soporte de estudios de imagen, como la angiografía con fluoresceína y/o indocianina, así como la tomografía óptica de coherencia de dominio espectral (HD-OCT).
Correspondencia: Dr. Alejandro Rodríguez García.
Instituto de Oftalmología y Ciencias Visuales, S.C. Centro Médico Zambrano-Hellion. Batallón de San
Patricio No. 112 Col. Real San Agustín. C.P. 66278. San Pedro Garza García, Nuevo León.
Teléfono: (81) 8676 2363, 8356 1878. Fax: (81) 8356 1799.
Correo electrónico: arodri@itesm.mx
