







Las Enfermedades del Tejido Conectivo (ETC) son entidades de baja prevalencia en la población general. Son de naturaleza inflamatoria y autoinmune, tienden a la cronicidad y al compromiso de muchos parénquimas, órganos y tejidos, dejando en ellos daño estructural y funcional. Dado lo anterior, amenazan la vida o disminuyen la expectativa y calidad de vida. El diagnóstico y tratamiento precoz de estas entidades, permite cambiar su curso y muchas veces lograr remisión. Es por lo tanto de suma importancia tenerlas en mente y sospecharlas como entidades de enfermedad e iniciar un tratamiento oportuno.
Connective Tissue Diseases have a low prevalence in the general population. They are inflammatory autoimmune diseases, chronic in nature and compromise different tissues and organs, leaving permanent and irreversible damage. They threaten live, and diminish quality and expectancy of life. Early diagnosis and treatment can change their natural course and in many cases induce remission. A high suspicion is necessary for a prompt diagnosis.
Las Enfermedades del Tejido Conectivo (ETC), también llamadas Enfermedades del Mesénquima o Mesenquimopatías, son entidades de baja prevalencia en la población general. Son de naturaleza inflamatoria y autoinmune, tienden a la cronicidad, y al compromiso de muchos parénquimas, órganos y tejidos, dejando en ellos daño estructural y funcional de variada cuantía. Dado lo anterior, amenazan la vida, o disminuyen la expectativa y calidad de vida. El diagnóstico y tratamiento precoz de estas entidades, permite cambiar su curso y muchas veces lograr remisión. Es por lo tanto de suma importancia tenerlas en mente y sospecharlas como entidades de enfermedad e iniciar un tratamiento oportuno.
Las más reconocidas son: Artritis Reumatoidea (AR), Lupus Eritematoso Sistémico (LES), Síndrome de Sjögren (SS), Esclerosis Sistémica Progresiva (ESP), Polimiositis (PM) y Dermatomiositis (DM), Enfermedad Mixta del Tejido Conectivo (EMTC). El Síndrome Antifosfolípidos (SAFL), enfermedad autoinmune, pero no de carácter inflamatorio se asocia frecuentemente a ellas, especialmente al LES.
Revisaremos con más profundidad LES y AR, donde ya se ha demostrado que el tratamiento en fases iniciales de enfermedad y un control estricto de la actividad de la enfermedad puede cambiar su evolución natural.
¿Cuándo sospechar una enfermedad del tejido conectivo?La clave para el diagnóstico parte con la historia clínica, primero escuchando atentamente las molestias del paciente, después interrogando sobre las características específicas de los síntomas. Esto sumado a un cuidadoso examen físico nos permite con alta probabilidad una opinión diagnóstica (1). Se solicitarán, en fase posterior, exámenes de laboratorio y/o imágenes para comprobarlo.
Los síntomas más frecuentemente relatados por los pacientes son: el dolor articular, aumento de volumen, rigidez, debilidad y/o falta de fuerza, fatiga y compromiso de estado general (Tabla 1). Es importante considerar la localización y la evolución de los síntomas, así como la respuesta a las posibles medicaciones instauradas previamente.
Muchas enfermedades reumatológicas se caracterizan por patrones específicos de dolor y rigidez. En las ETC, que son de carácter inflamatorio y sistémico, el dolor y rigidez tienden ser generalizados, y durar horas, peores al final del día y en la mañana. Ésto a diferencia del dolor mecánico, que tiende a ser localizado y regional y se asocia al sobreuso de una articulación afectada. Otros patrones específicos de dolor es el de la gota, que es de comienzo nocturno, agudo y quemante y suele afectar el primer ortejo; o el de la artrosis de manos, que da dolor y rigidez matinal de escasos minutos y desaparece después de iniciado el movimiento.
El aumento de volumen articular contado por los pacientes, es un síntoma muchas veces subjetivo e inespecífico y debe ser confirmado por signos objetivos de inflamación como aumento de temperatura, calor o enrojecimiento articular, sumada a la disminución de la movilidad y dolor. Estos caracterizan a las artritis. El modo de comienzo de la artritis (aguda o insidiosa), la duración de ésta (autolimitada o crónica), el número de articulaciones inflamadas (mono-oligo o poliartritis), la secuencia del compromiso articular (aditiva, intermitente o migratoria), su simetría o asimetría y su distribución (axial, periférica o ambas), permiten distinguir patrones muchas veces característicos de enfermedad. Entre las artritis agudas están aquellas inducidas principalmente por virus (parvovirus B19, rubeola) y bacterias (fiebre reumática, artritis reactivas), pero también pueden ser por cristales (gota o pseudogota). La AR tiene en general una forma de comienzo insidiosa, pero puede también debutar como aguda especialmente en personas mayores. En cuanto al número de articulaciones inflamadas es de importancia recalcar que en una monoartritis aguda hay que excluir las causas infecciosas y cristales principalmente y es de máxima utilidad la aspiración de la articulación afectada y el estudio del líquido sinovial. Las monoartritis crónicas pueden tener muchas etiologías: infecciosas (micobacterias, hongos), inflamatorias (cristales, psoriática, espondiloartritis anquilosante), asociada a enfermedades inflamatorias intestinales, cuerpos extraños, sarcoidosis, artrosis, tumores y otros. Las oligoartritis pueden tener las mismas etiologías que las monoartritis. Destacan en las oligoartritis el compromiso asimétrico de tobillo y rodilla o dolor sacroilíaco muy sugerente de una artritis reactiva; y el compromiso asimétrico de las pequeñas articulaciones de manos principalmente de IFD, sugerente de artropatía psoriática. El compromiso poliarticular y simétrico caracteriza a AR, artritis crónica juvenil, enfermedad de Still y al LES; en AR tiene gran predilección por las pequeñas articulaciones de manos, especialmente las metacarpofalángicas e IFP. El compromiso poliarticular asimétrico es característico de las pelviespondilopatías y artritis reactivas, en que también hay predilección por el compromiso axial (columna, sacroilíacas, esternoclavicular, esternal). Las artritis aditivas son características de las osteoartritis u artrosis, las artritis reactivas y la AR. Entre las artritis migratorias hay que pensar en fiebre reumática aguda y artritis por neisseria. Entre las artritis intermitentes pensar en artritis por cristales, enfermedad de Still y fiebre mediterránea familiar. Estos patrones de compromiso articular no son en sí diagnósticos, pero sí una gran guía para plantear un estudio más dirigido que nos permita confirmar nuestra hipótesis diagnóstica.
La debilidad o falta de fuerza es un síntoma común que puede tener muchos significados y debe ser analizado. Puede reflejar los efectos constitucionales de una enfermedad inflamatoria crónica o deberse a compromiso muscular (miopatías de diverso origen y polimiositis) o neurológico (neuropatías diversas). La debilidad muscular puede ser proximal, distal o localizada de comienzo agudo o insidioso y estar reflejando distinta patologías. La debilidad muscular por enfermedad inflamatoria muscular generalmente es progresiva en semanas o meses (al igual que la miastenia gravis, las distrofias musculares); pero también puede presentarse en forma aguda donde hay que diferenciarla de Guillan- Barre. En la polimiositis (PM) es simétrica y proximal, progresiva e invalidante y se debe al compromiso muscular inflamatorio. Los pacientes pueden relatar debilidad o falta de fuerza secundaria a muchas otras causas como el compromiso inflamatorio de una articulación, que les resta movilidad e incapacita para labores habituales.
La fatiga es una manifestación importante de la mayoría de las ETC principalmente AR y LES, donde es un buen indicador de la actividad de la enfermedad. Sin embargo la fatiga se encuentra en otras condiciones de base no inflamatoria como la fibromialgia.
Los paciente con enfermedades autoinmunes inflamatorias se pueden presentar con variados otros síntomas, que muchas veces no son relatados en forma espontánea y hay que preguntar en forma dirigida (Tabla 2). La sequedad de las mucosas, ocular y bucal se ven el Síndrome de Sjögren (S.S.), puede ser percibida como irritación ocular frecuente y a veces ojo rojo, y la necesidad de tomar mucha agua. La parotidomegalia recurrente también puede ser causada por SS. La alopecía difusa y la fotosensibilidad especialmente en área malar y escote caracteriza al LES. Las úlceras bucales pueden encontrarse en el LES, en espondiloartropatías seronegativas y en el Behcet. El fenómeno de Raynaud es característico en la Esclerodermia, pero puede encontrarse en otras ETC. Los fenómenos trombóticos y la pérdida recurrente del embarazo son característicos en el Síndrome Antifosfolípidos, que puede darse sólo o acompañar al LES. Siempre es importante preguntar por compromiso del estado general, como fiebre y baja de peso, que pueden tener muchas etiologías entre ellas las inflamatorias.
Sospecha de enfermedad de tejido conectivo
| Síntomas que deben ser investigados en forma dirigida |
|---|
| Artralgias |
| Rigidez articular mayor de 30 minutos |
Artritis: número de articulaciones comprometidas
|
| Falta de fuerzas: proximal o distal. |
| Sequedad de mucosa: xeroftalmía, xerostomía |
| Parotidomegalia recurrente |
| Alopecia |
| Fotosensibilidad |
| Úlceras, aftas mucosas |
| Raynaud |
| Tombosis |
| Muerte fetal in útero o pérdida recurrente de embarazo |
| Síndrome febril |
| Baja de peso |
En suma, los síntomas relatados por los pacientes en que hay que sospechar ETC son variados y subjetivos, deben ser precisados por el médico, quien además debe hacer un interrogatorio y examen físico más dirigidos para objetivarlos. Solo un alto índice de sospecha por permitirá en este punto hacer un diagnóstico precoz.
Artritis reumatoidea (AR)Es una de las ETC más frecuentes. Su prevalencia entre 0.5 y 1% en poblaciones europeas y americanas. La prevalencia es más baja en China y Japón (0.2-0.3%) y más alta en la población nativa norteamericana alcanzando un 5%.
Se define como una enfermedad inflamatoria autoinmune crónica que afecta las sinoviales articulares, y que en su evolución natural lleva a erosiones articulares llevando a destrucción progresiva e incapacidad funcional. Hay síntomas sistémicos y extra articulares en 40% de casos. El diagnóstico precoz y tratamiento temprano y agresivo puede cambiar el curso natural de la enfermedad, e inducir remisión (3).
Las alteraciones inmunológicas que llevan a la AR aparecen mucho antes de la enfermedad clínica. Esto es seguido de la aparición de síntomas o signos músculo esqueléticos vagos, que no permiten un diagnóstico, hasta que la respuesta inflamatoria sea de una magnitud suficiente para provocar inflamación sinovial y síntomas sistémicos. En estas primeras manifestaciones de la enfermedad estos pacientes pueden ser clasificados como artritis indiferenciada. Se han hecho grandes esfuerzo para detectar a estos paciente en esta fase precoz de enfermedad y establecer parámetros predictivos para progresión a AR definida. En forma paralela, se han cambiado los criterios de clasificación de la AR (7), lo que ha permitido incorporar a un número importante de pacientes en las fases iniciales de enfermedad para poder realizar estudios clínicos y terapias antes de que se produzcan lesiones irreversibles.
Formas de presentaciónLa AR afecta más a mujeres que hombres (2:1). Puede comenzar a cualquier edad, teniendo su máxima incidencia en la cuarta y quinta década de vida (2).
Tiene variadas formas de presentación y sintomatología que puede durar semanas o meses.
La forma más típica de presentación es de dolor articular difuso y simétrico y aumento de volumen que afecta las pequeñas articulaciones de manos, con dificultad para hacer un puño y rigidez matinal progresiva de más de 30 minutos o de horas. En las manos afecta las articulaciones metacarpofalángicas, interfalángicas proximales, muñecas, puede afectar tobillos y metatarsofalángicas. Este comienzo gradual se ve en el 50% de casos.
Mucho menos frecuente es la presentación monoarticular que puede afectar hombros o rodillas, seguido en los próximos días o semanas por una poliartritis aditiva que afecta muñecas, manos, tobillos o pies de manera generalizada.
La presentación aguda se da en 10 - 25% de casos. Puede presentarse como una poliartritis aguda con compromiso de hombros, codos, muñecas, manos, caderas, rodillas tobillos y pies. Este cuadro es muy doloroso e incapacitante, y aunque puede presentarse a cualquier edad, es más frecuente en el adulto mayor. Existe una forma de presentación que se caracteriza por poliartritis aguda de horas o días de duración intercalados por periodos sin síntomas. Estos episodios pueden recurrir en semanas o meses, pero se hacen más frecuentes y severos y evolucionan a una poliartritis persistente. La presentación como monoartritis es rara. Las tenosinovitis, bursitis y nódulos subcutáneos en las superficies extensoras pueden verse en asociación a las artritis, al igual que el síndrome del túnel carpiano, por compresión del nervio mediano, a nivel de la muñeca, por la articulación inflamada.
El adulto mayor puede presentarse con un cuadro de polimialgia reumática: poliartralgias y polimialgias que afectan cuello, hombros cadera y rodillas, asociada a fatiga y parámetros inflamatorios.
ProgresiónIndependiente de la forma de presentación, la progresión en AR puede tener varios patrones (3). Puede tener un curso breve y episódico, es decir un ciclo de enfermedad aguda seguida por remisión de al menos 1 año, lo que se da en 20% de pacientes. Puede tener un curso prolongado y progresivo de compromiso articular, en 10% de casos. Lo más frecuente es tener un curso de actividad intermitente o policíclico en 70% de casos. La severidad en estos episodios puede ser variable, pero la enfermedad activa lleva a mayor daño articular e incapacidad progresiva.
En la AR prolongada y activa hay frecuentemente síntomas sistémicos y compromiso extraarticular lo que es de peor pronóstico. La presencia de vasculitis, compromiso pulmonar y amiloidosis agrava el pronóstico. La seropositividad para factor reumatoide (FR) y anticuerpos anti péptido C citrulinado (APCC) se asocian a enfermedad más agresiva.
Los síntomas constitucionales como la fatiga y pérdida de peso pueden dominar el cuadro inicial. Los nódulos reumatoides se ven e en el 20% de AR activas seropositivas, y enfermedades más severa. Tienden a localizarse en áreas de presión como codos, articulaciones de manos y tendones aquilianos. Tienden a regresar a medida que la enfermedad se inactiva, pero pueden aumentar con el uso de ciertos medicamentos como el metotrexato. La anemia y trombocitosis son frecuentes en la enfermedad activa. La anemia es normocítica normocrómica, característica de la inflamación crónica. Ambas se normalizan en la enfermedad inactiva. La trombocitopenia es rara, se ve en el síndrome de Felty, que se define como AR en asociación con leucopenia y esplenomegalia; se puede observar en AR seropositiva, de larga data y agresiva. Puede haber elevación de enzimas hepáticas en enfermedad activa, como también este puede reflejar efecto tóxico de tratamientos antiinflamatorios o inmunosupresores usadas en la enfermedad.
El compromiso pulmonar en la AR es frecuente. En estudios de autopsia la pleura está comprometida en 50% de casos, puede pasar clínicamente inadvertida y ser asintomático. El derrame pleural es un transudado, su persistencia puede llevar a fibrosis. Pueden encontrarse nódulos pulmonares, que tienden a ser periféricos, es importante hacer diagnóstico diferencial con neoplasia, tuberculosis e infecciones por hongos. Lo más frecuente (28%) es el compromiso intersticial pulmonar difuso que puede llevar a fibrosis, es más frecuente en hombres que mujeres y enfermedad de larga evolución y seropositiva. El BOOP (neumonía organizada con bronquiolitis obliterante) tiene un mejor pronóstico y responde a corticoides.
El compromiso cardíaco en AR es el resultado de varios mecanismos de acción, como vasculitis, formación de nódulos, serositis, compromiso valvular y compromiso de vasos coronarios por ateromatosis.
El compromiso ocular más frecuente en la AR es la queratoconjuntivitis sicca (50%), el más grave y temido la escleritis que puede progresar a la escleromalacia.
En el compromiso neurológico en la AR, son frecuentes las neuropatías por atrapamiento, que no se asocian con severidad ni evolución de la enfermedad, sino con la inflamación sinovial articular que ejerce compresión sobre el nervio; los más frecuentes neuropatías compresivas del nervio mediano, cubital, tibial posterior y radial. La mielopatía cervical, la más temida, se debe a compresión medular secundaria subluxación atlantoaxoidea (SAA), se puede manifestar por Babinski e hiperreflexia. Dado su gravedad, la SAA debe ser buscada en forma dirigida con radiografías dinámicas de columna cervical.
El compromiso muscular en AR puede ser secundario a atrofia muscular por desuso, problemas nutricionales, efecto tóxico de tratamiento o neurológico. El compromiso renal no es frecuente en AR, pero puede ocurrir. Se describe nefropatía membranosa, glomerulitis, vasculitis y amiloidosis secundaria en AR activa de larga data.
La vasculitis de pequeño vaso es un fenómeno frecuente en la AR, y que puede explicar muchas de las manifestaciones e la AR. Puede comprometer la piel, manifestándose como infarto cutáneos (frecuentemente digitales), y úlceras en extremidades. También son frecuentes las neuropatías sensitivomotoras secundarias a vasculitis. La vasculitis en AR se asocia a enfermedad activa, un título elevado de FR, consumo de complemento, crioglobulinas y complejos inmunes circulantes, así como anemia y trombocitosis.
Fisiopatología de AREl concepto actual (4,5) presume que gatillos ambientales, presentados en forma de péptidos, interactúan con genes principalmente del complejo mayor de histocompatibilidad, estimulando inicialmente la respuesta inmune innata y después la respuesta inmune adaptativa. Es de gran importancia la generación de anticuerpos, como APCC (anticuerpos anti péptido citrulinado cíclico) y factores reumatoides (FR), que pueden preceder por años la aparición clínica de enfermedad. En la respuesta inmune adaptativa se produce la inflamación sinovial por la llegada de componentes del sistema inmune y células inflamatorias, creando una inflamación persistente, que finalmente erosiona al hueso.
A los factores genéticos se les atribuye un riesgo de 50% para desarrollar AR. Se han identificado más de 30 regiones génicas, las más estudiadas son HLA y PTPN22. Alelos HLA, como HLA-DRB1 contienen ciertas secuencias idénticas, lo que ha sido llamado “epítope compartido”. Se postula que los antígenos involucrados en la AR, antígenos artritogénicos, son modificados por un proceso llamado de citrulinación. Estos antígenos citrulinados son reconocidos por los alelos HLA que tienen este epítope compartido, quebrando la tolerancia y permitiendo la formación de anticuerpos contra ellos. Hay sin embargo, pacientes con AR que no producen APCC. Se considera entonces que la AR, es un síndrome clínico, formado por un grupo heterogéneo en lo genético y serológico, lo que puede manifestarse en variabilidad y pronóstico de la enfermedad. Alrededor de 50-80% de pacientes con AR tienen FR, APCC o ambos. Los APCC son predictores de peor pronóstico en AR, mayor daño articular y menor tasa de remisión de enfermedad (6).
En la AR hay distintas vías inflamatorias sobreexpresadas, como sobreproducción de Factor de Necrosis Tumoral (TNF), interleuquina 1, 6 y 17. Median comunicación entre células de la respuesta inmune y células de la inflamación, favorecen cascadas inflamatorias e inflamación sinovial. Algunos sinoviocitos (tipo macrófagos), pueden producir grandes cantidades de estas citoquinas proinflamatorias, otras sinoviocitos (tipo fibroblastos) tienen la capacidad de invadir el cartílago y migrar a otras articulaciones. Los osteoclastos activados en este ambiente inflamatorio pueden llevar a la erosión de hueso vecino.
Entre los factores ambientales asociados a AR, destaca el tabaquismo, que aumenta al doble el riesgo de desarrollar AR. Otros potenciales factores de riesgo son el consumo de alcohol, disminución de vitamina D, periodontitis y bajo nivel socioeconómico.
Criterios de clasificación ARLos clásicos Criterios del Colegio Americano de Reumatología de 1987 (ACR 1987), diseñados para distinguir AR de otras enfermedades del tejido conectivo y que cursan con artritis, identifican a pacientes con enfermedad ya establecida y erosiva. Dado la necesidad de identificar a pacientes con AR temprana (menor de 6 meses de evolución) antes de que exista enfermedad erosiva, se han creado nuevos criterios (ACR/ EULAR 2010) (7) (Tabla 3). Estos últimos documentan: 1- compromiso articular, 2- presencia y título de FR y APCC, 3- reactantes de fase aguda PCR y VHS y 4- duración de síntomas, dando una puntuación a las alteraciones encontradas. Para clasificar hay que tener una puntuación de 6 puntos o más de un total de 10 (8). Estos criterios tienen una sensibilidad de 73% y especificidad de 71%. Los criterios ACR 1987 tienen una sensibilidad de 47% y especificidad de 92% (9). La incorporación de Anti PCC aumenta la sensibilidad de los nuevos criterios, principalmente en sujetos con menor de 6 meses de evolución de enfermedad (10). Los criterios de clasificación no son criterios diagnósticos de enfermedad, son ideados para agrupar un grupo homogéneo de pacientes para poder comparar estudios, y observar medidas de intervención terapéutica.
Criterios acr/eular 2010
La población a aplicar estos criterios es aquella con
|
| 1 Compromiso articular (0-5) |
|---|
| Una articulación mediana o grande (0) |
| Dos a diez articulaciones medianas o grandes (1) |
| Una a tres pequeñas articulaciones (2) |
| Cuatro a diez pequeñas articulaciones (3) |
| Más de diez articulaciones (al menos una pequeña) (5) |
| 2 Serología (0-3) |
|---|
| Fr y APCC negativos (0) |
| FR o APCC positivos en títulos bajos (2) |
| FR o APCC positivos en títulos elevados (3) |
| 3 Reactantes de fase aguda (0-1) |
|---|
| PCR y VHS normal (0) |
| PCR o VHS elevada (1) |
| 4 Duración de síntomas (0-1) |
|---|
| Menos de 6 semanas (0) |
| Seis semanas o más (1) |
| Deben tener 6 puntos o más para ser clasificados como AR. Estos criterios permiten clasificar a aquellos pacientes que se presentan con sinovitis por primera vez. También pueden ser clasificados como AR, a pacientes con erosiones típicas de AR o AR de larga data que previamente cumplió criterios aunque este inactivo. |
Diagnosticar AR en sus primeros estadios es algo difícil. Muchos pacientes se presentan con una artritis indiferenciada y de duración menor de 12 semanas. Estos pacientes tienen sinovitis detectada al examen físico, sin encontrase causas como infección, enfermedad por cristales, espondiloartropatias o AR. La artritis indiferenciada es un diagnóstico de exclusión (11). La frecuencia de la artritis indiferencia va a depender de la duración de los síntomas: a mayor duración es mayor la probabilidad de realizar un diagnóstico definitivo. En este grupo de artritis indiferenciada, muchos remiten o se vuelven asintomáticos, pero la presencia de APCC da una posibilidad de 70% a desarrollar AR después de 1 año de evolución.
Se han identificado factores de riesgo independientes, para progresar a AR (11,12):
- 1-
Edad: La incidencia de AR es edad dependiente: en pacientes entre 18 y 34 años la incidencia es de 7/100.000; asciende a 34-107/100.000 entre los 75 a 84 años. Por lo que a mayor edad, mayor riesgo de desarrollar AR.
- 2-
Sexo. El sexo femenino que se presenta con artritis indiferenciada, tiene el doble de posibilidades de desarrollar AR.
- 3-
Número de articulaciones comprometidas y Proteína C Reactiva (PCR). El mayor número de articulaciones comprometidas con artritis y PCR elevada reflejan inflamación y se asocian con progresión de la sinovitis y peor pronóstico. Los pacientes que se presentan con poliartritis (más de 4 articulaciones comprometidas) tienen 1,5 veces más chance de desarrollar AR. Los que tienen más de 10 articulaciones inflamadas tienen 3 veces más riesgo.
- 4-
Autoanticuerpos. El FR y APCC pueden estar presentes años antes de los primeros síntomas clínicos de sinovitis. Son considerados factores de riesgo para desarrollar una sinovitis persistente y erosiva.
- 5-
Factores ambientales. Pacientes con artritis indiferenciada y que fuman tienen más riesgo de desarrollar AR y enfermedad más destructiva. Esto se ha observado en pacientes con alelos HLA-DRB1 y epítope compartido. También hacen con mayor frecuencia APPCC.
- 6-
Factores genéticos. Los factores claramente definidos son HLA-DRB1 y epitopes compartidos.
- 7-
Nuevos biomarcadores. En AR establecida los niveles de pro-MMP3, RANKL y osteoprotegerina se asocian con destrucción articular. Su significado es menos claro en la artritis indiferenciada.
Ventana de oportunidad para el tratamiento precoz Se define como el período temprano en el curso de la enfermedad cuando el proceso de enfermedad puede ser alterado o revertido. Se han intentado diferentes terapias en artritis indiferenciadas(13-17): corticoides, metotrexato, biológicos, con resultados alentadores como menor progresión a AR, menor progresión radiográfica, menor necesidad de uso de drogas modificadores de enfermedad. Pero como la artritis indiferenciada puede tener un curso variable y remisión espontánea en 40-50% (18,19), la decisión de tratar o no debe ser personalizada y recaer en el especialista, dado el efecto tóxico de drogas potenciales a usar.
El tratamiento de la AR establecida debe ser lo más precoz posible y enérgico, siendo la meta la remisión de enfermedad. Se emplean los llamados “fármacos modificadores de enfermedad” (FARMEs) o DMARDs en inglés (Disease Modifying anti Rheumatic Drugs) solos o en combinaciones unos con otros, con corticoides en la etapa inicuial; si estos no logran controlar la enfermedad se agrega tratamiento biológico. El Metotrexato es el primer FARMe usado en AR, y debe ser indicado en el diagnóstico de AR, en dosis progresivas. Si estuviera contraindicado se puede usar Leflunomida o Sulfazalazina. Si no hay respuesta a la monoterapia se pueden asociar FARMEs o combinar con tratamiento biológico. Los anti-TNF generalmente son usados en primera línea en la actualidad, pero otros han mostrado eficacia como los anti CD20, anti IL6 y moduladores selectivos de la co-estimulación entre célula presentadora y linfocito T, como abatacept.
Lupus eritematoso sistémico (LES)El LES, dada su naturaleza de enfermedad crónica, autoinmune e inflamatoria que puede afectar a cualquier órgano de la economía, es un desafío diagnóstico para el no especialista, ya que su presentación puede ser bastante heterogénea. Como puede ser una enfermedad grave y fatal, es importante sospecharla, hacer un diagnóstico precoz e iniciar terapéutica lo antes posible. El paciente lúpico debe ser controlado en forma regular, para prevenir o tratar en forma oportuna las exacerbaciones de la enfermedad y prevenir el daño irreversible de órganos como riñón, pulmón, corazón o sistema nervioso.
El LES es una enfermedad que afecta a minorías. La prevalencia es variable en distintas series y depende del sexo, grupo de edad, raza y características demográficas. Es de 14-78 casos por 100.000 adultos mayores de 18 años (20). Es más frecuente en afroamericanos y en el sexo femenino (10:1). La gran mayoría son mujeres en edad reproductiva, pero puede presentarse a cualquier edad, tanto en niños como adultos mayores.
Presentaciones clínicasLa presentación (21, 22) más típica es la de una mujer joven con compromiso del estado general, artralgias u artritis, puede tener fiebre y adenopatías, úlceras orales, caída del cabello y lesiones cutáneas diversas como el desarrollo de eritema en las zonas foto expuestas a la luz solar, siendo lo más característico el eritema sobre las mejillas y el dorso de la nariz. Puede a veces encontrarse hipertensión arterial, orinas espumosas u oscuras, dolor torácico tipo pleurítico y a veces dolor abdominal.
Sin embargo no siempre nos enfrentaremos a la presentación típica, puede tratarse de un niño o de un adulto mayor o de un varón, y/o debutar con síntomas de mayor gravedad como compromiso de conciencia y/o accidente cerebrovascular, convulsiones o psicosis; o con compromiso neurológico medular o periférico aislado, síndrome nefrítico o nefrótico, equimosis por trombocitopenia o anemia hemolítica.
La presentación clínica puede ser distinta en diferentes grupos etarios. En niños es más frecuente debutar con compromiso agudo y grave como el renal, encefalopatía o anemia hemolítica si lo comparamos con adulto (23). El LES que debuta en el adulto mayor (10-20%) de casos, puede dar síntomas inespecíficos de curso larvado, lo que retrasa el diagnóstico, son menos frecuentes las manifestaciones cutáneas, la fotosensibilidad, la artritis y la nefritis aguda, y más frecuentes la serositis, el compromiso pulmonar, fiebre, síndrome de Raynaud y Ssicca.
También es necesario pensar en la posibilidad de LES en mujeres con pérdida reproductiva que desarrollan compromisos de estados general o síndromes inflamatorios posterior al desenlace de su embarazo, o flebitis. Entre las alteraciones de laboratorio que nos hagan plantear un LES, especial mención al VDRL falsamente positivo (con FTABs o RPR negativo), la anemia crónica, la anemia hemolítica, la leucopenia y/o linfopenia persistentes, la plaquetopenia, la hipoalbuminemia con proteinuria y/o hematuria, especialmente con dismorfia y/o con cilindros hemáticos, y el compromiso renal rápidamente progresivo, entre otros.
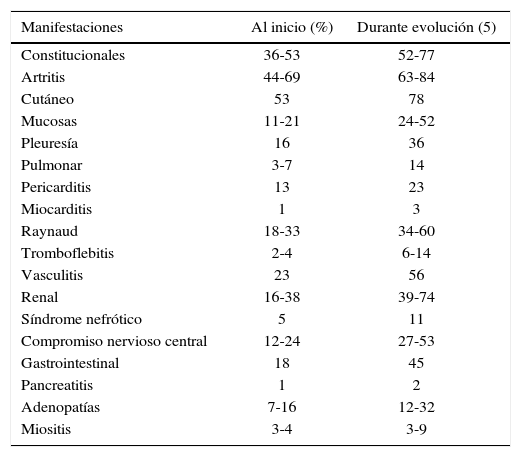
En suma, el LES puede tener una presentación bastante heterogénea, y caracterizarse inicialmente por síntomas inespecíficos. La frecuencia de síntomas al inicio de la enfermedad y durante la evolución de ésta, de dos grandes series de pacientes (Toronto con 376 pacientes y Europea con 1000 pacientes) se resumen a continuación en la siguiente Tabla: (modificada de Rheumatology, second Edition. John H. Klippel, Paul A. Dieppe. Systemic Lups Erythematosus: Clinical features. (Capítulo 7, pág 1;2).
| Manifestaciones | Al inicio (%) | Durante evolución (5) |
|---|---|---|
| Constitucionales | 36-53 | 52-77 |
| Artritis | 44-69 | 63-84 |
| Cutáneo | 53 | 78 |
| Mucosas | 11-21 | 24-52 |
| Pleuresía | 16 | 36 |
| Pulmonar | 3-7 | 14 |
| Pericarditis | 13 | 23 |
| Miocarditis | 1 | 3 |
| Raynaud | 18-33 | 34-60 |
| Tromboflebitis | 2-4 | 6-14 |
| Vasculitis | 23 | 56 |
| Renal | 16-38 | 39-74 |
| Síndrome nefrótico | 5 | 11 |
| Compromiso nervioso central | 12-24 | 27-53 |
| Gastrointestinal | 18 | 45 |
| Pancreatitis | 1 | 2 |
| Adenopatías | 7-16 | 12-32 |
| Miositis | 3-4 | 3-9 |
La presentación clínica tan variable hace difícil el diagnóstico si no se tiene un alto grado de sospecha. Resulta muy útil preguntar en forma dirigida por: fotosensibilidad, caída del cabello, úlceras mucosas, preguntar si hay parientes en la familia con LES u otra enfermedad del Tejido Conectivo. Hay que realizar un examen físico cuidadoso y solicitar un estudio de laboratorio pertinente.
Entre los exámenes de mayor utilidad en el diagnóstico están: 1- hemograma (en busca de anemia crónica o anemia de carácter hemolítico, leucopenia, linfopenia, plaquetopenia), 2- complemento C3 y C4 (disminuidos en enfermedad activa), 3- orina completa (para evidenciar proteinuria, hematuria, leucocituria, o cilindros) y 4- anticuerpos antinucleares (ANA). Estos anticuerpos están presentes hasta en un 99% de pacientes activos sin tratamiento, por lo que son de mucha utilidad ante la sospecha inicial. Pero los ANA son bastante inespecíficos y no son exclusivos del LES, pueden estar presentes en otras condiciones reumatológicas o en enfermedades infecciosas, o post vacunación, pero su presencia en el contexto clínico antes mencionado da mayor probabilidad al diagnóstico. Si están positivos se deben solicitar anticuerpos más específicos para LES, como anti DNAn y anti ENA, como el anti Sm, ya que ambos son marcadores específicos de esta enfermedad.
El LES es un desafío diagnóstico para el no experto y puede simular otras condiciones con las cuales hay que hacer diagnóstico diferencial. Las enfermedades infecciosas son una posibilidad en aquellos que se presentan con fiebre, adenopatías y rash como la mononucleosis infecciosa, infección por Parvovirus B19, HIV. Pero también hepatitis virales si hay compromiso hepático, o endocarditis bacteriana si hay soplos cardíacos o lesiones sugerentes de vasculitis cutáneas. Muchas condiciones se pueden presentar con compromiso del estado general y dolor articular y/o muscular, entre ellas la fibromialgia, los síndromes paraneoplásicos, o presentarse como artritis franca en la que hay plantear artritis virales, reactivas y o otras enfermedades del tejido conectivo.
Criterios de clasificación de LESEl Colegio Americano de Reumatología (ACR) ha definido criterios para la clasificación de LES (24), para poder comparar pacientes de distintos centros y además distinguirlos de otras enfermedades del tejido conectivo. En 1997 estos fueron reevaluados y se incluyó la presencia de anticuerpos antifosfolípidos (25). El síndrome antifosfolípido se puede encontrar en casi la mitad de pacientes con LES y da morbilidad agregada que se caracteriza por eventos trombóticos recurrentes y abortos repetidos y/o muerte fetal in útero en las pacientes lúpicas que se embarazan.
Estos son:
- 1.
Rash malar: eritema fijo, plano u elevado sobre las mejillas, respetando pliegue nasogeniano.
- 2.
Rash discoide: placa o placas eritematosas solevantadas, con queratosis y clavos córneos.
- 3.
Fotosensibilidad: Rash cutáneo secundario a exposición solar.
- 4.
Ulceras orales: ulceraciones orales o nasofaríngeas, generalmente indoloras.
- 5.
Artritis: no erosiva, de 2 o más articulaciones periféricas.
- 6.
Serositis:
- a:
Pleuritis
- b:
Pericarditis
- a:
- 7.
Renal:
- a:
Proteinuria persistente de más de 0.5gr/día o más de 3.
- b:
Cilindros celulares (Hemáticos, granulares, tubulares o mixtos).
- a:
- 8.
Neurológico
- 9.
Hematológico
- a:
Anemia Hemolítica con reticulocitosis, o
- b:
Leucopenia: < de 4.000cel/mm3 en 2 o más ocasiones, o
- c:
Linfopenia: < de 1.500cel/mm3 en 2 o más ocasiones, o
- d:
Trombocitopenia: < de100.000/mm en ausencia de posibles drogas.
- a:
- 10.
Inmunológico
- a:
Anticuerpos contra DNA nativo, o
- b:
Anticuerpos anti Sm, o
- c:
Anticuerpos Antifosfolípidos: Anticardiolipinas o Anticoagulante Lúpico o VDRL falsamente positivo (con test treponémico negativos) y persistente al menos por 6 meses.
- a:
- 11.
Anticuerpos antinucleares.
La presencia de 4 o más criterios, sea en forma simultánea o en el curso de la evolución de la enfermedad, permite clasificar al paciente como LES. A la luz de los conocimientos actuales y con la intención de que estos criterios de clasificación incluyan a pacientes en el inicio de su enfermedad, o que no hayan complicado los 4 criterios, estos criterios se están reevaluando (26). En la actualidad los pacientes que no cumplen con 4 criterios de clasificación son llamados LES probable, LES-like o Enfermedad indiferenciada del Tejido Conectivo.
Morbilidad en LESLa historia natural del LES se caracteriza por episodios de recaídas o de actividad, que alternan con períodos de remisión o inactividad; pero es altamente variable habiendo pacientes que pueden remitir por años, o tener poca expresión clínica de enfermedad y otros morir por la enfermedad activa o sus complicaciones. Tanto la morbilidad como la mortalidad ha mejorado a lo largo de los años: en 1955 la mortalidad era de 50% a 5 años, en la actualidad 93% sobrevive a los 5 años y 83% está vivo a los 10 años. Se cree inciden en estas cifras una indicación más cautelosa de corticoides, terapias inmunosupresoras, manejo de infecciones, control y prevención de complicaciones a largo plazo como la ateroesclerosis generalizada.
Los corticoides, usados para control de enfermedad, tienen efectos adversos a largo plazo, que hay que predecir, monitorear y evitar. Su uso se asocia a hiperlipemia, aumento de presión arterial, ganancia de peso e hiperglicemia. A mayor plazo se asocian con osteoporosis y osteonecrosis, enfermedad cardiovascular y cataratas. Si se planea usar corticoides por largo plazo, debe usarse una segunda droga para lograr controlar la enfermedad y minimizar y/o lograr suspender los corticoides. La hidroxicloroquina es frecuentemente usada para este propósito y se piensa en la actualidad que debe ser usada en todo paciente lúpico. Se ha asociado con disminución de episodios de actividad, se cree previene daño neurológico y renal, tiene acción antitrombótica y mejora la respuesta a micofenolato de mofetilo (27). La vigilancia renal es de suma importancia en el paciente lúpico. La nefritis lúpica, no diagnosticada y tratada a tiempo puede llevar a daño renal irreversible. Se debe solicitar orina completa y función renal en forma regular y si aparece proteinuria, un buen índice de seguimiento es la relación microalbuminuria/ creatininuria en orina aislada. Si se compromete la función renal o si hay proteinuria > de 500mg se debe realizar una biopsia renal, para documentar el tipo de daño renal, el grado de inflamación o de cronicidad de las lesiones e instaurar la terapéutica más adecuada.
Las reactivaciones en LES pueden ser de variada gravedad, puede haber compromiso cutáneo y artritis, como manifestación de activación de enfermedad. Pero pueden haber compromiso de órganos nobles como riñón, pulmón (hemorragia pulmonar), tejido nervioso (cerebritis, mielitis transversa), hematológico (anemia hemolítica) o trombótico (Sindrome antifosfolípido catastrófico) que son verdaderas emergencias y deben ser tratadas en forma enérgica. Los pacientes lúpicos tienen hasta 50% mayor riesgo de trombosis si tiene presencia de anticuerpos antifosfólipidos, principalmente anticoagulante lúpico. Este riesgo puede ser reducido con el uso de aspirina e hidroxicloroquina.
El embarazo en LES es otro tema de importancia, ya que estos paciente no tienen infertilidad, pero sí mal pronóstico de su LES y de su embarazo. Este mal pronóstico se asocia a actividad al momento de embarazo o si el período de inactividad pre-embarazo ha sido menos de 6 meses, sobre todo para la nefritis lúpica, que se activa durante el embarazo y en el postparto. La presencia de anticuerpos antifosfolípidos se asocia con pérdida recurrente de embarazo, con muerte fetal in útero, trombosis y trombocitopenia; la presencia de anticuerpos anti ENA Ro y La se asocian con LES neonatal y bloqueo cardíaco congénito dado que atraviesan la barrera placentaria. La presencia de anticuerpos antitiroideos se asociada con pérdida de embarazo y parto prematuro en paciente lúpicas. Todos estos autoanticuerpos deben ser controlados en pacientes que planean o están en curso de su embarazo.
Una de las causas principales de muerte en LES es de origen cardiovascular (10%). La ateroesclerosis se puede encontrar en 50% de pacientes lúpicos. Los factores de riesgo cardiovascular tradicionales (obesidad, hiperlipemia, tabaquismo entre otros) deben ser monitoreados y tratados.
FisiopatologíaSe piensa que la etiología del LES es multifactorial, interviniendo factores genéticos, inmunológicos, hormonales, ambientales, e infeccioso que conjugados unos con otros lleva a la pérdida de tolerancia inmunológica y la aparición de autoreactividad patológica (28). Se sabe que los anticuerpos Antinucleares (ANA), anti Ro y anti DNA pueden preceder por años la aparición de la enfermedad clínica (29).
Hay más de 30 genes descritos (30) en asociación con LES, la mayoría de asociados a la respuesta inmune y la inflamación. Se piensa que es una enfermedad poligénica, aunque se conoce que la deficiencia aislada de C1Q lleva a una pobre eliminación de células apoptóticas, y que la deficiencia de C4 se asocia a una menor eliminación de linfocitos B autoreactivos (28). Este riesgo genético puede estar influenciado por efectos epigenéticos como la poca metilación del DNA o modificaciones en histonas, que se pueden heredar o ser secundarias a factores ambientales. Como dijimos, el LES tiene una fase preclínica caracterizada por la presencia de autoanticuerpos. Se piensa que factores ambientales como drogas, exposición solar, stress y/o infección son los factores precipitantes para la progresión y la aparición de manifestaciones clínicas de enfermedad. Hay un proceso de amplificación de la enfermedad, donde los ácidos nucleicos de las células apoptóticas (auto-antígenos) estimulan la producción de interferón alfa y promueve la enfermedad activando a las células presentadoras de antígeno. Existe una mala eliminación y/o degradación de células apoptóticas, por lo que estos autoantígenos circulan por más tiempo, facilitando la perpetuación de la reactividad inmunológica. Los complejos inmunes formados se pueden depositar en varios órganos, activando complemento, donde reclutan células inflamatorias que contribuyen al daño. En el sitio de la inflamación hay producción de citoquinas proinflamatorias y actividad procoagulante (31) lo que amplifica el daño.
TratamientoLas estrategias terapéuticas apuntan a múltiples objetivos (31, 32): capacidad de diagnóstico precoz, definiciones de actividad o exacerbaciones, estratificación de acuerdo a severidad del compromiso, uso de terapias para inducir remisión precoz y prevenir exacerbaciones, prevención y manejo de comorbilidades. No es la intención de este artículo abarcar todos estos aspectos. Las terapias más frecuentemente usada son los corticoides, hidroxicloroquina, azatioprina, metotrexato, micofenolato mofetil y ciclofosfamida. Existe en uso terapias biológicas como Rituximab (antiCD20) y Belimumab (anti Blyss) (33).
En suma, dada la baja prevalencia de estas enfermedades, pero su alta morbilidad, hay que estar atentos a reconocerlas desde sus inicios. El diagnóstico oportuno y un tratamiento eficaz y agresivo intentando lograr remisión de la enfermedad, permitirían evitar los daños orgánicos que ocurren en la evolución natural de la enfermedad. Los síntomas de presentación pueden ser inespecíficos, mas un interrogatorio dirigido y un examen físico cuidadoso pueden orientar a la posibilidad de una enfermedad del tejido conectivo. El apoyo del laboratorio en indiscutible en el diagnóstico precoz, la presencia Fr y aPCC en artritis de corta evolución apoya el diagnóstico de AR. La presencia de ANA, anti ENAs (Sm, Rnp) y antiDNAn, sumado a complementos bajos entre otros apoyan diagnóstico de LES.
¿CuÁndo se debe sospechar LES?
| 1 Mujer en edad reproductiva, con compromiso de estado general, artralgias y/o artritis. |
| 2 Fiebre de origen desconocido con o sin poliadenopatías. |
| 3 Alteraciones cutáneas y mucosas: Fotosensibilidad, eritema malar, caída del cabello, vasculitis, úlceras mucosas y lesiones cutáneas crónicas y fijas. |
| 4 Serositis: pleuritis, pericarditis. |
| 5 Convulsiones o Psicosis de origen reciente, asociado a alteraciones sistémicas como compromiso estado general, o de alteraciones de laboratorio como anemia, leucopenia, linfopenia, o alteración del sedimento de orina. |
| 6 Mujer embarazada o post parto, con compromiso de estado general, artralgia o artritis, o flebotrombosis o parto prematuro por estas condiciones. |
| 7 En cuadros de anemia hemolítica o trombocitopenia o leucopenia o linfopenia persistentes; en el estudio de anemias crónicas. |
| 8 En pacientes con VDRL persistentes positivos, sin evidencias de sífilis. |
| 9 En pacientes de cualquier edad con anemia normocítica normocrómica, de carácter persistente. |
La autora declara no tener conflictos de interés, con relación a este artículo.