



Las Enfermedades Neuromusculares representan un grupo heterogéneo de trastornos que incluyen alteraciones en la motoneurona, nervios periféricos, transmisión neuromuscular y las patologías que afectan al músculo propiamente tal. Su progresión varía considerablemente presentando diversos déficits que pueden variar desde la debilidad muscular, pérdida sensorial, dolor, fatiga y la disfunción autonómica, los que se combinan para dar lugar al daño músculo-esquelético generando limitaciones en las actividades de la vida diaria y restricciones en la participación. Es por esto, que en las últimas décadas se han creado programas de rehabilitación integrados por un equipo multidisciplinario, que trabajan de manera coordinada, abarcando todos los factores que producen y aumentan la discapacidad realizando el control, la prevención y el tratamiento de las complicaciones que derivan durante la evolución de este grupo de enfermedades, con el objetivo de lograr la mayor integración que el estadio de su enfermedad pueda permitir.
Neuromuscular Diseases constituted an heterogeneous group of disorders that can include motor neuron alterations, peripheral nerves, neuromuscular transmission and pathologies that affect the muscle itself. Its progression varies considerably, presenting as various deficits that can vary from muscle weakness, sensory loss, pain, fatigue and autonomic dysfunction. All of this can combine and result in musculoskeletal injury, causing limitations in daily living activities and restrictions in participation. It is for this reason that in recent decades integrated rehabilitation programs have been created, conform by a multidisciplinary team, working in a coordinated manner, encompassing all factors that produce and increase disability, achieving the monitoring, prevention and the treatment of complications that can arise during the evolution of this group of diseases, with the goal of achieving the greater integration that their disease stage allows.
Las enfermedades neuromusculares (ENM) se definen como un grupo de enfermedades que afectan alguno de los componentes de la unidad motora: célula del hasta anterior, nervio periférico, unión neuromuscular y/o músculo. Se consideran enfermedades raras, por su baja incidencia y prevalencia.
Son de naturaleza hereditaria o adquirida y pueden presentarse en cualquier etapa de la vida; en la infancia y adolescencia el origen es generalmente genético.
Se caracterizan por la debilidad muscular, generalmente progresiva, lo que conlleva a una disminución en su capacidad funcional global, comprometiendo otros sistemas y produciendo una dependencia cada vez mayor (1).
Las ENM en su gran mayoría no tienen tratamiento etiológico, son incurables pero no intratables, de esta manera, la rehabilitación es fundamental, y su objetivo es maximizar las capacidades funcionales, proporcionando una mejor integración comunitaria con una buena calidad de vida tanto al paciente como a su familia (2).
El programa de rehabilitación debe ser instaurado precozmente, ser constante, individualizado y adaptado a cada paciente según el estadio evolutivo. El equipo debe ser multidisciplinario, de esta manera se logra una mayor efectividad en los resultados.
El propósito fundamental de este artículo es, por lo tanto, constituir un aporte desde el área de la rehabilitación en el abordaje de tres patologías que se originan en diferentes estructuras de esta Unidad Motora, seleccionadas por su prevalencia, por requerir de un abordaje multidisciplinario y por las necesidades que presentan en términos de rehabilitación: Atrofia Muscular Espinal (SMA por su sigla en inglés), Distrofia Muscular de Duchenne (DMD) y Charcot Marie Tooth (CMT).
2Atrofia muscular espinal (SMA)La SMA es una enfermedad de herencia autosómica recesiva, caracterizada por la degeneración de las neuronas motoras en la médula espinal, generando una debilidad muscular proximal progresiva. La forma clásica de la enfermedad es causada por una mutación en el gen SMN1 localizado en 5q11.2-q13.3, afectando la supervivencia de las neuronas motoras. Existe un gen homólogo, llamado SMN2, cuya presencia es necesaria para la sobrevida de estos niños, por lo que el número de copias de SMN2 podría correlacionarse con la gravedad del fenotipo. Se distinguen cuatro formas de presentación en función de la edad de inicio y de la función motora máxima alcanzada. Se agrega un tipo 0 (forma severa, con síntomas antenatales). La frecuencia de portadores es 1/ 54 y la incidencia 1/11.000 (3). Es la segunda causa de muerte en la infancia por enfermedad hereditaria recesiva (4). El diagnóstico es principalmente clínico y la confirmación es a través del estudio genético identificando la deleción del gen SMN1 en el cromosoma 7 (95% de sensibilidad y 100% de especificidad).
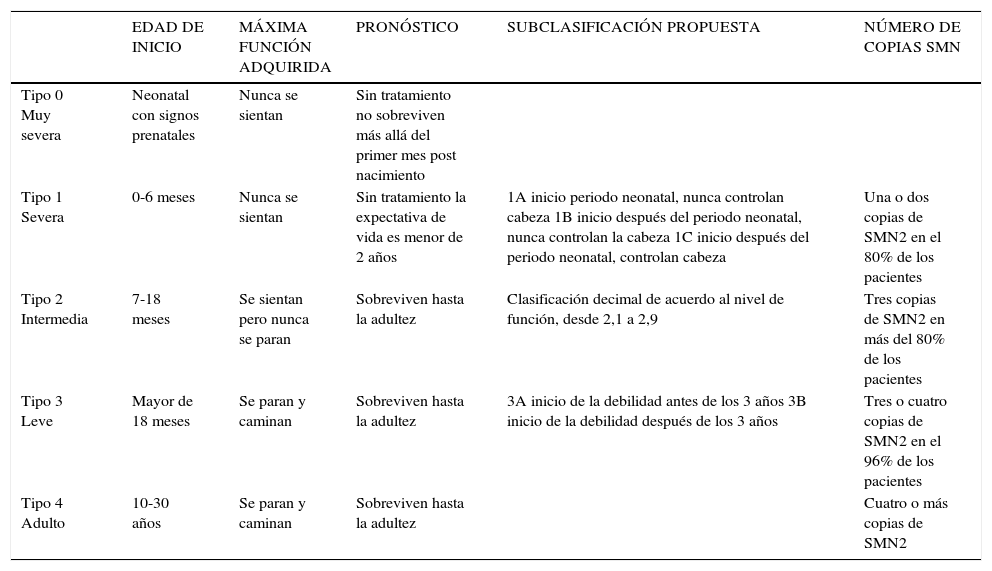
En SMA tipo I, el inicio suele ser antes de los 6 meses, no logran el sedente sin apoyo, y la sobrevida menor a 2 años. El SMA tipo II, se inicia entre los 6 y los 18 meses, logran sentarse de forma independiente, pero no ponerse de pie o caminar sin ayudas y la sobrevida es hasta la edad adulta. En SMA tipo III, el inicio es posterior a los 18 meses, logran estar de pie o caminar sin ayuda, pero pueden perder este hito a una edad más tardía, la sobrevida es esencialmente normal. El tipo IV aparece desde la segunda década de vida (Tabla 1).
Clasificación de la atrofia muscular espinal
| EDAD DE INICIO | MÁXIMA FUNCIÓN ADQUIRIDA | PRONÓSTICO | SUBCLASIFICACIÓN PROPUESTA | NÚMERO DE COPIAS SMN | |
|---|---|---|---|---|---|
| Tipo 0 Muy severa | Neonatal con signos prenatales | Nunca se sientan | Sin tratamiento no sobreviven más allá del primer mes post nacimiento | ||
| Tipo 1 Severa | 0-6 meses | Nunca se sientan | Sin tratamiento la expectativa de vida es menor de 2 años | 1A inicio periodo neonatal, nunca controlan cabeza 1B inicio después del periodo neonatal, nunca controlan la cabeza 1C inicio después del periodo neonatal, controlan cabeza | Una o dos copias de SMN2 en el 80% de los pacientes |
| Tipo 2 Intermedia | 7-18 meses | Se sientan pero nunca se paran | Sobreviven hasta la adultez | Clasificación decimal de acuerdo al nivel de función, desde 2,1 a 2,9 | Tres copias de SMN2 en más del 80% de los pacientes |
| Tipo 3 Leve | Mayor de 18 meses | Se paran y caminan | Sobreviven hasta la adultez | 3A inicio de la debilidad antes de los 3 años 3B inicio de la debilidad después de los 3 años | Tres o cuatro copias de SMN2 en el 96% de los pacientes |
| Tipo 4 Adulto | 10-30 años | Se paran y caminan | Sobreviven hasta la adultez | Cuatro o más copias de SMN2 |
En todos los tipos de SMA, los músculos proximales son más débiles que los distales. Esta debilidad es simétrica y compromete los miembros inferiores antes que los superiores. En las extremidades superiores, se compromete en primer lugar los músculos de la cintura escapular, con mayor afectación de los grupos extensores sobre los flexores. Sin embargo, en el cuello predomina el compromiso de los grupos flexores y en el tronco no existe diferencias entre ambos grupos (5).
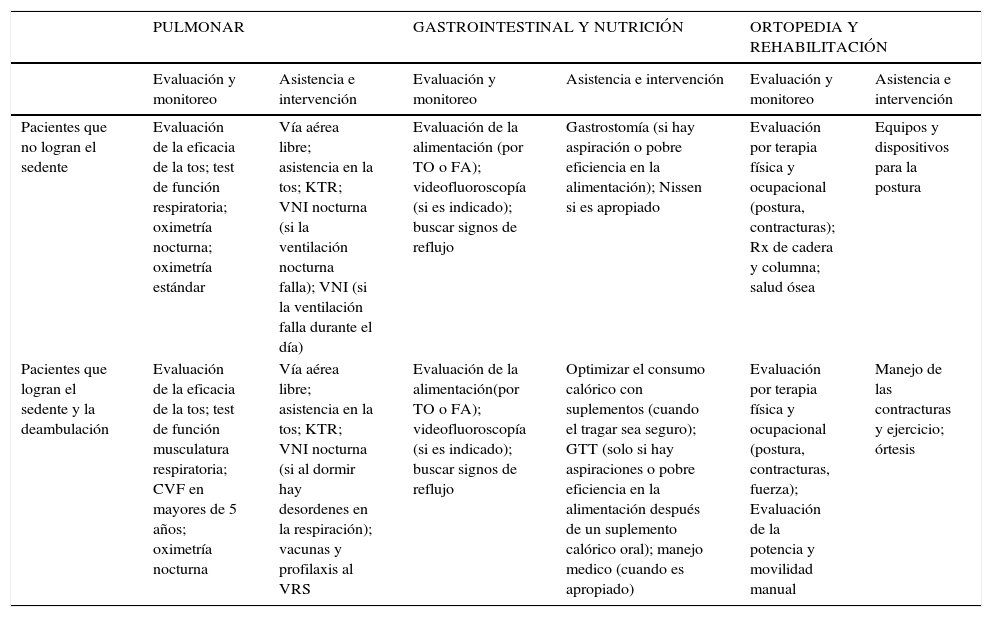
Los cuidados en SMA, fueron estandarizados por un comité de expertos, trabajo publicado en el año 2007 como una declaración de consenso en relación a normas de atención (3). Tabla 2.
Cuidados Sma Estandari Zados
| PULMONAR | GASTROINTESTINAL Y NUTRICIÓN | ORTOPEDIA Y REHABILITACIÓN | ||||
|---|---|---|---|---|---|---|
| Evaluación y monitoreo | Asistencia e intervención | Evaluación y monitoreo | Asistencia e intervención | Evaluación y monitoreo | Asistencia e intervención | |
| Pacientes que no logran el sedente | Evaluación de la eficacia de la tos; test de función respiratoria; oximetría nocturna; oximetría estándar | Vía aérea libre; asistencia en la tos; KTR; VNI nocturna (si la ventilación nocturna falla); VNI (si la ventilación falla durante el día) | Evaluación de la alimentación (por TO o FA); videofluoroscopía (si es indicado); buscar signos de reflujo | Gastrostomía (si hay aspiración o pobre eficiencia en la alimentación); Nissen si es apropiado | Evaluación por terapia física y ocupacional (postura, contracturas); Rx de cadera y columna; salud ósea | Equipos y dispositivos para la postura |
| Pacientes que logran el sedente y la deambulación | Evaluación de la eficacia de la tos; test de función musculatura respiratoria; CVF en mayores de 5 años; oximetría nocturna | Vía aérea libre; asistencia en la tos; KTR; VNI nocturna (si al dormir hay desordenes en la respiración); vacunas y profilaxis al VRS | Evaluación de la alimentación(por TO o FA); videofluoroscopía (si es indicado); buscar signos de reflujo | Optimizar el consumo calórico con suplementos (cuando el tragar sea seguro); GTT (solo si hay aspiraciones o pobre eficiencia en la alimentación después de un suplemento calórico oral); manejo medico (cuando es apropiado) | Evaluación por terapia física y ocupacional (postura, contracturas, fuerza); Evaluación de la potencia y movilidad manual | Manejo de las contracturas y ejercicio; órtesis |
Existen diferentes escalas para realizar una evaluación funcional: Hammersmith Functional Motor Scale for Spinal Muscular Atrophy, y Medición de la Función Motora (MFM) (6), usadas para objetivar déficit, planificar estrategias de abordaje y mejorar el seguimiento.
Tratamientoa)Prevención de contracturas y manejo posturalLas contracturas constituyen una complicación constante, debido a la gran debilidad muscular y se instauran más precozmente a mayor severidad del cuadro. Se observa en más de la mitad de los pacientes y se relacionan con periodos de inactividad. Contracturas superiores a 45° se consideran intratables (6). Suelen ser deformidades en flexión y aparecen inicialmente en las extremidades inferiores. Para algunos autores son inevitables, no obstante pueden retrasarse si se instaura un tratamiento rehabilitador precoz (4)
SMA tipo I Movilización de cada una de las articulaciones en todo su rango, si existen contracturas mandibulares, apoyo de fonoaudiología. Cambios frecuentes en los diferentes decúbitos. Asegurar un sedente estable y confortable con diferentes opciones de asientos moldeados, los que pueden ser incluidos en la silla de ruedas. Ortesis de diferentes tipos para prevención de contracturas y dolor en casos de hipermovilidad (6). Terapia de juego utilizando juguetes livianos. Uso de soportes para antebrazos con el objetivo de facilitar los movimientos en el plano transverso y asistencia tecnológica disponible.
SMA tipo II la debilidad muscular, contracturas, escoliosis y alteraciones respiratorias son los principales problemas. Elongaciones diarias y cambios en diferentes decúbitos, especialmente prono son fundamentales. Ortesis son indicadas con el objetivo de preservar rangos articulares, palmetas de reposos en extremidades superiores y ortesis tobillo pie (AFO). Con el objetivo de apoyar la bipedestación en mobiliarios adecuados, se usan ortesis rodilla tobillo pie (KAFO). En casos especiales, marcha con ortesis reciprocas. El manejo postural debe incluir mobiliario de sedestación a la medida y silla de rueda que permita la máxima independencia. La bipedestación con ortesis debe ser realizada durante 2 a 3 horas diarias fraccionadas (6), ya que, además de la prevención de contracturas en EEII, contribuye a mejorar la densidad ósea a nivel de la columna vertebral y el fémur (7).
SMA tipo III los principales problemas se dan por la dificulta para realizar las transiciones entre el suelo, sedente y bípedo, caminar distancias mayores, o en terrenos irregulares, subir escalas y caídas frecuentes generadas por la debilidad muscular proximal y trastornos en el balance (equilibrio). Se quejan de fatiga, deformidades músculo esqueléticas y dolor. Las fracturas secundarias a las caídas no son raras. (6) En este grupo, durante el período de marcha, las contracturas son casi inexistentes. Una vez perdida la marcha, el abordaje es semejante al grupo anterior. Uso de silla de ruedas para distancias largas, en forma paralela al fomento y mantención de la marcha en espacios más reducidos
b)EscoliosisSe presenta entre un 78 a 100% (4). Hay predominancia de curvas torácicas y tóraco lumbares derechas y lumbares izquierdas (6). Suelen ser curvas flexibles, que pueden acompañarse en menor frecuencia de cifosis. Iniciada la escoliosis, evoluciona de forma rápida. En el tipo II su inicio es precoz, con una edad media de cuatro años. En el tipo III, la aparición es más tardía y menos constante y suele coincidir con la etapa de pérdida de marcha (4). Los corsé pueden ser utilizados para soporte postural, pero no hay pruebas suficientes para apoyar el retraso en la progresión de la curva. Si se utilizan deben ser fabricados con un recorte abdominal, permitiendo la adecuada excursión diafragmática y acceso a la gastrostomía si la tiene (6). En el tipo II, se suele indicar corsé o asientos moldeados con el objetivo de proporcionar una mayor estabilidad del tronco en el sedente, permitir un uso funcional de sus manos y retrasar la cirugía. (4) El tratamiento quirúrgico es el manejo de elección, pero no hay consenso en relación a la edad para realizarlo, como tampoco del rol de las ortesis espinales en su manejo (3). La corrección quirúrgica de la escoliosis debe ser considerada sobre la base de progresión de la curva, la función pulmonar y madurez ósea. La cirugía de escoliosis en niños con prolongada supervivencia proporciona beneficios en el equilibrio sentado, resistencia y cosmesis (6).
c)Luxación de caderasAunque frecuente, es rara vez doloroso (6) y se relaciona con la ausencia de carga. Es más frecuente en niños que no realizan marcha. Es controversial si la postura inadecuada en sedente generando una oblicuidad pélvica, contribuiría a una subluxación o luxación. Para algunos autores la luxación de cadera es un factor desencadenante de escoliosis, para otros, ambos se desarrollarían paralelamente como consecuencia de la gran debilidad de los músculos de la cintura pélvica y tronco. La reducción quirúrgica es controversial debido a un elevado porcentaje de recidivas, la que estaría condicionada por la gran debilidad muscular pelviana. Se debe establecer un tratamiento preventivo con fisioterapia, control postural en sedente, prolongando al máximo, el período de bi-pedestación y marcha.
d)Alteraciones de la marchaSMA tipo III todos los niños alcanzan la marcha aunque su inicio suele ser tardío. Pueden tener dificultad para correr y subir escaleras. Alrededor de la mitad perderán la deambulación independiente cerca de los 14 años (3). Esta pérdida puede ocurrir a otras edades, solo una pequeña fracción son deambuladores durante toda la vida (4). La indicación de silla de ruedas depende de factores como la frecuencia de caídas y fatiga (3). Desarrollan estrategias para caminar, diferentes a los pacientes portadores de DMD a pesar de tener similares debilidades musculares.
Mantienen la estabilidad limitando los momentos articulares y usando la rotación pélvica para impulsar la pierna hacia adelante. Presentan una amplia base de sustentación, con abducción de cadera, la fuerza muscular en el glúteo medio tiene un alto valor predictivo de la marcha. Presentan una inclinación de tronco hacia atrás para compensar la debilidad de los extensores de cadera. Los movimientos compensatorios de brazo, tronco y cabeza son esenciales en el plano frontal. Las estrategias terapéuticas deben propender a mantener la máxima potencia de los flexores de cadera y abductores (8).
e)Manejo RespiratorioLa enfermedad pulmonar es la principal causa de morbimortalidad en la SMA I y II y en menor grado en el tipo III. El compromiso es de los músculos inspiratorios y espiratorios con predominio de estos últimos, estando el diafragma relativamente preservado. (6) En el tipo I, además de la importante debilidad muscular, se observa un retraso en la maduración pulmonar con un menor número de alvéolos en el nacimiento, el resultado es una caja torácica en forma de campana con depresión esternal. Los principales problemas respiratorios son: Alteración de la tos, generando una pobre eliminación de las secreciones de las vías respiratorias bajas. Hipoventilación durante el sueño. Menor desarrollo de la caja torácica y pulmonar. Infecciones recurrentes que agravan la debilidad muscular.
Sin asistencia respiratoria los niños con SMA tipo I fallecen antes de cumplir los 2 años. En el tipo II estos trastornos se producen en menor frecuencia, pero son también la principal causa de morbimortalidad. En el tipo III, las complicaciones respiratorias son más raras. El manejo de estas complicaciones debe realizarse en forma preventiva, desde el momento del diagnóstico. Las evaluaciones van desde el examen físico hasta la polisonmografia.
En todos los pacientes es vital tener una vía aérea libre y una forma de lograrlo es asistiendo en la tos, manual o con máquinas y aspiración posterior, con drenaje de secreciones, a través de kinesiterapia respiratoria. Manejo adecuado y oportuno de las infecciones agudas del tracto respiratorio superior. El soporte ventilatorio en pacientes con hipercapnia diurna (4,6), disminuye los síntomas de trastornos respiratorios durante el sueño y mejora la calidad de vida.
f)EjercicioEstudios en modelos de ratones con SMA, que imitan la forma humana, demuestran que el ejercicio físico es beneficioso, evidenciado por un aumento significativo en la sobrevida y las capacidades motrices. (9,10) El ejercicio no solo tiene un efecto sobre el músculo, sino también sobre el sistema nervioso autónomo, regulando la función cardíaca y respiratoria.
Se refuerza la importancia de las elongaciones, el posicionamiento adecuado, uso ortésico, bipedestación y marcha para prevenir o retrasar la aparición de contracturas. (11,12)
Los ejercicios de estiramiento o elongaciones consisten en llevar el músculo a su longitud máxima a través de la articulación afectada, manteniéndola adecuadamente alineada y estabilizando las articulaciones que no se están moviendo, al lograr el rango completo de movimiento, mantener durante al menos 10 segundos y luego repetir. No hay evidencia suficiente para establecer la frecuencia y duración de este tipo de ejercicios. (13)
Ejercicios de fortalecimiento muscular de esfuerzo submáximo, se consideran seguro y convenientes ya que evitan la atrofia por desuso, especialmente en patologías lentamente progresivas. El objetivo es mantener la fuerza existente o retardar la progresión de la debilidad, no para fortalecer los músculos debilitados.(14)
El ejercicio aeróbico como natación, de intensidad moderada, es beneficioso para disminuir la obesidad, mejorar el estado cardiorrespiratorio, otorgar mayor sensación de bienestar y favorecer el aumento de la masa ósea. (14)
Las recomendaciones por grupo de expertos son: (15,16)
- 1
Adoptar un estilo de vida activo, por los beneficios físicos y psicológicos
- 2
Elongación y movilizaciones pasivas como manejo preventivo de las contracturas
- 3
Ejercicio aeróbico moderado y ejercicio de resistencia moderada
- 4
Evitar ejercicios de alta intensidad
Los pacientes con SMA no logran alcanzar independencia en AVD acorde a su edad, el objetivo será proporcionar herramientas para lograr la mayor autonomía posible. La incapacidad para movilizar brazos contra gravedad, limita actividades como higiene, vestuario y alimentación. Los soportes móviles de antebrazos anclados a las sillas de ruedas facilitan estas actividades. La escritura es mantenida hasta fases avanzadas, pueden coger un lápiz pero se agotan con rapidez, al igual que con otros elementos como cuchara, cepillo de dientes, etc. Estrategias como engrosar un lápiz o adaptaciones tipo cartucho contribuyen a mantener la independencia en estas áreas. Un PC es recomendado para facilitar la escritura y por lo tanto el trabajo escolar. Adaptación a la vivienda, introducción de sistemas de control del entorno (domótica). Silla de ruedas electrónica, si es incapaz de autopropulsar una mecánica, ayudan a mantener la integración escolar y social (4)
3Distrofia muscular de duchenne (DMD)La DMD es una enfermedad hereditaria de carácter recesivo ligada al X, que se presenta durante la infancia y que afecta aproximadamente 1 de cada 3500-6000 (17-19) varones nacidos vivos. Es la ENM más frecuente de la infancia (2). DMD se produce como resultado de mutaciones (principalmente deleciones) en el gen de la distrofina (locus Xp21.2). Las mutaciones pueden conducir a una ausencia o defecto de la distrofina, una de las principales proteínas que mantiene la estructura de la fibra muscular, resultando en la degeneración muscular progresiva. Se ha encontrado una alta incidencia de mutaciones de novo, ya que en 1/3 de los casos no se encuentran antecedentes familiares (19).
La primera manifestación clínica puede ser un retraso en el desarrollo psicomotor, retrasando la adquisición de la marcha independiente; posteriormente dificultad para correr, subir escalas y saltar debido a la gran debilidad muscular proximal, la que se evidencia con una maniobra de Gowers positiva. Este signo asociado a una hipertrofia de gemelos, es orientador al diagnóstico. Puede existir una disfunción cognitiva no progresiva, dada la presencia de distrofina en el cerebro (17). El retardo mental leve se ve en 1/3 de los pacientes (1). En algunos casos se puede observar un retraso en el lenguaje.
El diagnóstico suele realizarse alrededor de los 5 años (17). CPK muy elevada, biopsia muscular y estudio genético contribuye al diagnóstico de certeza. Es importante el estudio genético en las madres, para descartar su condición de portadoras, información que será útil para otras mujeres, miembros de la familia materna (hermanas, hijas).
Al progresar se genera una debilidad de los extensores de cadera y flexores dorsales de tobillo, llevando a una postura en hiperlordosis lumbar, amplia base de sustentación y apoyo en equino. Aumentan las caídas, la fatiga, hasta la pérdida de la marcha independiente entre los 7-12 años con un promedio de 9,5 (20). Con la pérdida de la deambulación aparecen nuevas complicaciones: escoliosis, contracturas articulares, atrofia por desuso, complicaciones respiratorias, cardiacas, nutricionales (desnutrición u obesidad), digestivas (constipación o reflujo gastroeso-fágico), osteoporosis y fracturas patológicas. Sin intervención, la edad promedio de muerte es alrededor de los 19 años por insuficiencia respiratoria (75%) o cardiaca (25%) (1,17,19). Existe una forma más leve y menos frecuente en la cual hay una disminución de la distrofina, la Distrofia Muscular de Becker DMB (1). Es una enfermedad similar, pero cuyas manifestaciones clínicas son menos graves y la edad de aparición es más tardía, por definición, los pacientes con DMB conservan la capacidad ambulatoria después de los 16 años (19).
TratamientoDebe enfocarse en cuatro aspectos fundamentales: mantener la fuerza muscular, prevenir deformidades de columna, manejar complicaciones respiratorias y prevenir y tratar alteraciones cardíacas.
Los corticoides han cambiado el curso de la DMD, son hasta el momento, el único tratamiento que ha demostrado enlentecer la progresión de la debilidad muscular reduciendo el riesgo de escoliosis y estabilizando la función pulmonar. Existe evidencia sólida que mejoran la fuerza y la función muscular a corto plazo (de 6 meses a 2 años).
Asociado a terapia física, sería la estrategia terapéutica actual de mayor éxito, ya que el tratamiento a largo plazo se relaciona con una disminución en el ritmo de deterioro de la fuerza muscular y función motora (17) y por ende de la capacidad ventilatoria y prolongación de la marcha independiente (hasta 3,3 años) v/s los que no lo usan (20), por lo tanto menor riesgo de desarrollar escoliosis y complicaciones respiratorias y cardíacas (19). Los efectos secundarios deben ser prevenidos y manejados precozmente. Se recomienda no iniciarlos mientras el paciente este ganando habilidades motoras. Su indicación será cuando las habilidades motrices ya no mejoran pero aún no han empezado a empeorar, esto ocurre entre los 4-6 años (21). No hay consenso a la edad en que deben ser retirados.
Etapa de deambulación- a)
Manejo postural: Las deformidades más frecuentes durante la etapa de deambulación son caderas en flexión y abducción, rodillas en flexión y pies en equino varo.
Los factores que contribuyen a la generación de contractura son: pérdida de la capacidad de mover en forma activa una articulación en todo su rango, posición estática en flexión, desbalance muscular y cambios fibróticos en el músculo.
Las elongaciones son una medida preventiva benéfica y debe ser parte de una rutina diaria.
Se recomienda elongar cada grupo muscular, mínimo 4 a 6 días a la semana, fundamentalmente cadera rodilla y tobillo.
Ortesis AFO no deben ser indicadas para la marcha, ya que al bloquear el mecanismo compensatorio, la impiden. Solo se indican en reposo, generalmente uso nocturno, con el objetivo de prevenir contracturas a nivel de tobillo. En etapas cercanas a la pérdida de la marcha se pueden agregar ortesis KAFO, para prevenir las contracturas a nivel de rodilla, apoyar en la bipedestación y eventualmente la marcha (17).
La edad de indicación de silla de ruedas no está bien establecida, se puede indicar una silla liviana, para trayectos mayores cuando estos superan la resistencia del paciente (18).
Las cirugías de alargamiento en extremidades inferiores son discutidas (17,22).
- b)
Columna: La aparición y evolución de las deformidades de columna están relacionadas con la fuerza y el tono muscular. A menor tono y fuerza, mayor posibilidad de desarrollar deformidades de columna, que aparezcan a menor edad y progresen más rápidamente. El seguimiento de la columna se realiza desde antes de que ocurra la pérdida de la marcha, ya que al perderla, la escoliosis comienza a desarrollarse. Se recomienda evaluación clínica de columna en cada control y radiografía si se observa curva escoliótica. (17,23).
- c)
Cuidados respiratorios: Mientras el paciente con DMD mantenga la marcha, las complicaciones respiratorias son escasas, la afectación y las complicaciones respiratorias se producen en una progresión gradual. Se recomienda realizar una evaluación de función pulmonar CVF anual, esto le permite al niño familiarizarse con el equipo y al tratante evaluar la función respiratoria máxima alcanzada.
- d)
Cuidados cardiológicos: Las alteraciones cardiológicas corresponden a Miocardiopatía secundaria a fibrosis y alteraciones del ritmo y la conducción (24). Las manifestaciones clínicas de una Insuficiencia Cardiaca son reconocidas tardíamente (17). Se ha demostrado que las lesiones aparecen a temprana edad y permanecen silentes durante años, por lo que la Academia Americana de Pediatría recomienda vigilar a los pacientes constantemente, para lograr la detección y el tratamiento oportuno. Desde el diagnóstico, las evaluaciones cardiológicas se realizan cada dos años hasta los 10 años, incluyendo como mínimo un electro y ecocardiograma. Posterior a esta edad o si los síntomas aparecen antes, los controles serán anuales, con evaluaciones cardiológicas completas (19,23).
- e)
Abordaje del lenguaje: Se ha observado en algunos niños con DMD déficits del habla y lenguaje, incluyendo memoria verbal a corto plazo, procesamiento fonológico, así como coeficiente intelectual disminuido y trastornos específicos del aprendizaje. Es importante la detección temprana y derivación oportuna (23).
- f)
Alteraciones de la marcha: Los pacientes con DMD desarrollan estrategias y/o mecanismos compensatorios destinados a mantener su marcha por más tiempo a pesar de la debilidad muscular en progresión. Estudios realizados en Laboratorio de Marcha evidencian en el plano sagital las principales compensaciones. Mantienen la posición bípeda usando el equino y la hiperlordosis lumbar, de esta manera compensan la debilidad de los extensores de cadera. En la marcha aparece una flexión de cadera, circunducción de la extremidad y flexión plantar del tobillo. La anteversión pélvica compensa la falta de extensión de la cadera. Las estrategias terapéuticas deben tratar de mantener la máxima potencia de los flexores de cadera y abductores. En DMD, la posición en equino parece proporcionar una ventaja para caminar, por lo cual es importante preservar la fuerza muscular de los flexores plantares (8). Sobre esta base, el alargamiento quirúrgico del Tendón de Aquiles es discutido, pues podría producir la pérdida de la marcha al suprimir el efecto compensatorio del equino.
- a)
Manejo postural: énfasis en la prevención del desarrollo de contracturas articulares y musculares, a través de las elongaciones y uso ortésico, incluyendo a las extremidades superiores. Si las contracturas articulares en extremidades inferiores lo permiten es posible mantener la bipedestación con ortesis tipo KAFO (17). Las cirugías en extremidades inferiores se pueden realizar en casos excepcionales.
Pueden presentar dolor de causa postural, se debe buscar una posición cómoda en sedente y en los diferentes decúbitos, y proporcionar el apoyo ortésico adecuado (23).
La indicación de silla de ruedas permanente se realizará cuando las caídas son frecuentes y presentan una dificultad extrema para levantarse desde la posición sedente, el buen posicionamiento es prioritario al realizar la prescripción, considerando la capacidad de auto propulsarla (18). Si sus condiciones motoras no lo permiten, la silla de ruedas electrónica es la mejor alternativa, con el fin de mantener la independencia en esta área (17).
En esta etapa hay mayor predisposición a fracturas por la movilidad disminuida, debilidad muscular y la terapia corticoidal, generando osteoporosis y dolor. El manejo farmacológico con calcio, vitamina D y bifosfonatos sumado a las terapias físicas se hace necesario (17).
- b)
Columna: sin corticoides, desarrollan escoliosis progresiva en un 90%. El uso de éstos ha demostrado reducir el riesgo o al menos retrasar su aparición.
La edad de pérdida de la deambulación se relaciona directamente con el desarrollo de deformidades de columna vertebral, por lo que es necesario realizar controles anuales con radiografía, si la curva es menor a 15 o 20 grados y cada 6 meses si es mayor de 20 grados (17). El abordaje preventivo es orientado a mantener una postura simétrica de columna y pelvis en el sedente.
Las terapias físicas y corsé no modifican la evolución natural de la escoliosis, los ejercicios tienen como objetivo evitar rigideces, manteniendo el rango de movilidad articular y el corsé tiene como objetivo mantener el tronco alineado y estable a la espera de un tratamiento definitivo (quirúrgico) en el momento oportuno, pero puede ser indicado si la cirugía no puede ser realizada o no es la opción elegida (23,22).
La cirugía de columna se indica cuando las funciones cardíaca y respiratoria no están gravemente afectadas. La edad óptima para operar es entre los 11 y 13 y preferentemente cuando el ángulo de Cobb está entre 20-40°. El objetivo es obtener y mantener el balance postural que le permita permanecer sentado el resto de su vida, evitando la deformación progresiva de columna, que restringe la capacidad ventilatoria (19).
- c)
Manejo respiratorio: pérdida la marcha se debe enfatizar en la evaluación pulmonar, realizando oximetría de pulso, CVF sentado, flujo máximo de tos, presión inspiratoria y espiratoria máxima (17). Con la progresión de la enfermedad se incluyen otras evaluaciones, como la polisomnografia.
Los efectos del entrenamiento de los músculos respiratorios son controvertidos, para algunos autores es benéfico ya que mantendrían la fuerza y preservarían la función pulmonar, mientras que otros sugieren que el entrenamiento de músculos respiratorios podría incrementar el daño. (19,25). Al progresar la debilidad de los músculos respiratorios aparecen complicaciones como tos inefectiva, hipoventilación nocturna, somnolencia diurna, cefalea matinal, trastornos respiratorios del sueño y por último falla respiratoria diurna. La tos asistida y la ventilación nocturna no invasiva está demostrado que aumentan la sobrevida. Las intervenciones dependerán de la fase de la enfermedad. En una primera etapa es útil aumentar la cantidad de aire que entra en los pulmones, mediante la respiración profunda (técnicas de inflación del pulmón). Con la progresión de la enfermedad, la tos se vuelve menos eficaz, y las técnicas manuales y mecánicas de asistencia para toser pueden ser muy útiles. Con el tiempo, el apoyo ventilatorio no invasivo nocturno y luego diurno será necesario (23,17,25).
- d)
Seguimiento cardiológico: similar al grupo anterior, dado que se incrementa el riesgo de problemas cardiacos.
- e)
Actividades de la vida diaria: al progresar la enfermedad, la fuerza de los brazos es cada vez menor, se recomiendan dispositivos de asistencia para mantener la independencia en AVD y participación. Adaptaciones a utensilios para comer o para higiene menor, uso de soporte móvil para antebrazos, ayudas técnicas que faciliten las transferencias, etc. Apoyo de tecnología asistida, de acuerdo a las necesidades. Indicación de silla de ruedas electrónicas si la mecánica no es capaz de autopropulsar.
- f)
Ejercicio en Distrofinopatías: existe discrepancia entre distintos autores en relación a las recomendaciones. Para algunos, los ejercicios de baja intensidad y los ejercicios aeróbicos de baja resistencia mantienen o mejoran ligeramente la fuerza muscular, mientras que otros señalan que los ejercicios inducen a la debilidad y al aumento de la tensión mecánica generando mayor daño muscular ya que, las contracciones excéntricas máximas dañan al citoesqueleto, ocasionando una transitoria debilidad muscular con aumento de los niveles de CK, produciendo dolor y acelerando la degeneración muscular (26,27).
Los ejercicios de fuerza contra una gran resistencia y los ejercicios excéntricos están contraindicados durante todas las etapas de la enfermedad porque pueden exacerbar el daño muscular. (17). Recordar que la inestabilidad inherente de la membrana del sarcolema con deficiencia de distrofina, predispone a lesiones debido a las cargas mecánicas (28). Los pacientes que padecen de DMD presentan una baja capacidad cardiovascular y una baja utilización periférica de oxígeno con una mayor frecuencia cardiaca en reposo además de desarrollar una insuficiencia cronotrópica, es decir, tienen una disminuida capacidad para aumentar su ritmo cardiaco en respuesta al ejercicio y en reposo presentan una frecuencia cardiaca de 110 ± 12 latidos/minuto. Por lo anterior, se ha demostrado que realizar ejercicio aeróbico de intensidad moderada, especialmente en etapas tempranas de la enfermedad, como piscina o bicicleta, mejora la capacidad aeróbica, por lo que se recomienda en fase ambulatoria este tipo de ejercicio sin superar el 20% de la contracción voluntaria máxima (fortalecimiento submáximo). Al ejecutarlos en forma suave, sin generar fatiga muscular, evitaría la atrofia por desuso. Se puede continuar realizándolos en fase no ambulatoria, siempre que sea medicamente seguro.
Esta recomendación es aplicable a pacientes con Distrofia muscular Miotónica, distrofia de cinturas, DMB y miopatías inflamatorias, donde puede mejorar la fuerza y la función, sin signos de aumento de la inflamación muscular (29-31). Las recomendaciones de consenso por grupo de expertos son similares a las señaladas para AME.
4Polineuropatías hereditarias HMSNLas Polineuropatías corresponden a trastornos del nervio periférico, que se manifiestan por compromiso motor y sensitivo, hipo o arreflexia y atrofia muscular de predominio distal. Comprende desde el punto de vista etiopatogénico tres grupos: polineuropatías hereditarias, adquiridas y las asociadas a trastornos neurodegenerativos (32).
Las neuropatías hereditarias incluyen una amplia serie de síndromes, sin embargo, las más frecuentes y de mayor prevalencia corresponde a la enfermedad de Charcot-Marie-Tooth (CMT) también conocida como neuropatía sensitivo-motora hereditaria HMNS (33).
El CMT es un síndrome polineuropático sensitivo-motor, desmielinizante o axonal, que puede transmitirse con herencia autosómica dominante, recesiva o ligada al X. Pese a su semiología estereotipada es genéticamente complejo, dado que se han localizado 36 loci con una treintena de genes mutantes clonados. Cualquiera que sea el defecto estructural que afecte a la mielina o al axón, la vía final común está representado por un proceso degenerativo axonal que explica el fenotipo típico. (34,35)
Combinando estudios clínicos, electrofisiológicos y la anatomía patológica de la biopsia del nervio, se han diferenciado en dos grandes tipos: un tipo desmielinizante (HMSN tipo I o CMT1, con velocidad de conducción inferior a 38 m/sg) originado por mutaciones en alguno de los varios genes que se expresan en las células de Schwann y un tipo neuroaxonal la HMSN tipo II o CMT2, con velocidad de conducción superior a 38 m/sg) cuya alteración primaria es el axón (32,33). Cada uno de estos grupos puede subdividirse en otros (36).
El CMT1 corresponde a la variedad más frecuente, le sigue la forma desmielinizante ligada al cromosoma X y luego CMT 2 (33,37).
Clínica y afectación musculoesqueléticaLos síntomas de inicio son la torpeza para correr y la dificultad para ponerse de talones. Los músculos del pie son los que primero se atrofian, dando lugar al pie cavo varo y dedos en garra producto del compromiso desigual de los diferentes grupos musculares, siendo la afectación del compartimiento lateral de la pierna el más importante, preservándose el compartimiento posterior hasta las fases finales de la enfermedad. La debilidad del músculo tibial anterior ocasiona una caída del pie por déficit de la musculatura extensora del tobillo, lo que provocaría la marcha en “steppage” (38). A medida que la enfermedad progresa, se pueden atrofiar los músculos de la pierna y el tercio inferior del muslo, dando lugar a las características “patas de cigüeña” o de “botella de champán invertida” por la pérdida de masa muscular. Con el tiempo se pueden afectar las manos, que se manifiestan con deformación progresiva y aplanamiento o atrofia de las eminencias tenar e hipotenar lo que genera dificultad para llevar a cabo habilidades motoras finas (34,37).
El inicio de la marcha es generalmente a edad normal y los síntomas comienzan en la infancia o adolescencia, aunque algunas formas pueden empezar en la edad adulta. En la mayoría de los casos los pacientes no se quejan de síntomas sensitivos, siendo la expresión más patente de la alteración de la sensibilidad y la inestabilidad producto de la pérdida de la propiocepción lo que puede incluso ocasionar una ataxia sensorial. El dolor espontáneo no lo manifiestan, pero si el dolor de pies, debido al mal apoyo, a las callosidades y a las deformidades de éstos. Los reflejos osteotendinosos se reducen o están ausentes (39).
La mayoría de los pacientes son totalmente independientes durante gran parte de su vida, pero hay algunos tipos que dan lugar a una sintomatología más grave. La progresión de la enfermedad en general es muy lenta, pero variable de una persona a otra, incluso dentro de los miembros de una misma familia, mostrando una lenta evolución en las últimas décadas de la vida (34,37).
Las alteraciones de cadera son escasas, aparecen con mayor frecuencia en el tipo I, preferentemente entre la 1° y 2° década de la vida (6-8%), cuando se ve afectada la musculatura proximal, comprometiendo a los abductores y extensores de cadera generando rotación externa, coxa valga, anteversión femoral, subluxación y displasia acetabular. En un comienzo, la patología de cadera es asintomática, posteriormente presenta dolor y alteraciones de la marcha, es importante tenerla en consideración solicitando un estudio radiológico en forma habitual. El manejo en esta etapa es quirúrgico (40)
Compromiso de otros órganos:La afectación respiratoria es rara en CMT, pero se ha informado en CMT1A, CMT2C, y otros tipos de CMT. La debilidad del diafragma, la parálisis de las cuerdas vocales en aducción, la apnea del sueño y el síndrome de piernas inquietas puede estar asociada con el CMT y requerir un tratamiento adecuado (41).
Abordaje de RehabilitaciónNo existe un tratamiento específico para cada tipo de CMT, por esto, la principal función del médico es el diagnóstico preciso y precoz de la enfermedad, para así iniciar un tratamiento sintomático y rehabilitador adecuado y oportuno (42).
Este tipo de enfermedades si bien no reduce la esperanza de vida, son evolutivas y progresivas a lo largo del tiempo, por lo que acompañaran al paciente durante toda su vida. La evolución no es predecible y el grado de discapacidad puede ir desde un pie cavo que requiera plantillas hasta un paciente que requiera la silla de ruedas para el traslado.
El tratamiento de estas patologías neuromusculares es multidisciplinario y se centra en los siguientes objetivos:
- a)
Mejorar fuerza muscular.
- b)
Prevenir las deformidades esqueléticas.
- c)
Mantener una adecuada marcha.
- d)
Evitar caídas.
- e)
Facilitar la habilidad manual.
- f)
Tratar el dolor.
- g)
Entregar apoyo psicológico.
- h)
Mantener funcionalidad.
Para el logro de estos objetivos el tratamiento se basa en lo siguiente:
- a)
Educación: Dirigida al paciente, familia y cuidador, explicando en que consiste la enfermedad, su condición de cronicidad, progreso y evolución poco predecible. Educar en cómo prevenir o retrasar complicaciones. Proporcionar un adecuado consejo genético al paciente y familia.
- b)
Terapia de Rehabilitación:
Kinesiterapia:
- -
Ejercicios de acondicionamiento general y fortalecimiento muscular de extremidades y tronco para la prevención de deformidades en forma precoz e individualizada principalmente con ejercicios de fortalecimiento de la musculatura intrínseca.
- -
Ejercicios de elongación y flexibilización, para evitar acortamientos tendíneos (Tendón aquiliano, fascia plantar) y retracciones articulares.
- -
Ejercicios que favorezcan la propiocepción y con ello mejorar el equilibrio y la marcha.
- -
Ejercicios aeróbicos para mejorar la funcionalidad y capacidad aeróbica.
Todas estas modalidades tendrán niveles de participación e intensidad variables que dependerán del grado de compromiso del paciente, el que puede oscilar desde ejercicios asistidos a ejercicios contra resistencia. Existen algunos ensayos clínicos aleatorizados que establecen como evidencia que el ejercicio, de leve a moderada intensidad, es efectivo y seguro para los pacientes con CMT y que conducen a una mejora significativa en la capacidad de caminar y en el fortalecimiento de los miembros inferiores. Es controversial si el ejercicio de alta intensidad genera mayor debilidad muscular, por lo cual, la recomendación es evitarlo (34).
Terapia Ocupacional: Facilitar las AVD que permitan la máxima funcionalidad e independencia. Mejorar o mantener las actividades que requieren manipulación fina, promover la higiene articular, evaluar la necesidad de ortesis en miembros superiores para prevenir deformidades, mantener posturas adecuadas y mejorar la posición funcional. Objetivar la necesidad de aditamentos y ayudas técnicas (bastón, carros andadores y otros) que permitan una mayor independencia y que disminuyan el riesgo de caídas prolongando la capacidad de marcha.
- -
- c)
Ortesis: El tratamiento de las deformidades de los pies depende de la edad del paciente, de la flexibilidad de éstos, de la deformidad ósea y el desequilibrio muscular. Una AFO de uso nocturno o de tiempo completo moldeada a la forma del pie, puede reducir la tendencia hacia un mayor desarrollo de la deformidad. Un pie flexible se puede manejar sin cirugía usando una AFO sólida en posición neutra. La atrofia muscular y la debilidad condicionan un pie equino-varo reductible en una primera etapa, que dificulta la deambulación, una AFO, estabiliza el pie, mejora el apoyo y favorece un mejor patrón de marcha al controlar la caída del antepie en fase de balanceo. Sin embargo, pocos ensayos aleatorios controlados han evaluado la eficacia de las órtesis, por lo que sea cual fuere la indicación siempre es necesario combinar su uso con ejercicios de elongación del pie asociado a la bipedestación y deambulación (43,44).
En una primera etapa, el pie cavo varo puede requerir de una plantilla con barra retrocapital y una cuña de base externa para controlar la supinación del retropié. En etapa posterior una ortesis de pie (FO), puede cumplir mejor esta función. Posteriormente la AFO es la mejor indicación. En etapas iniciales en que hay inestabilidad de tobillo, se debe indicar un calzado adecuado, deportivo o con caña alta. Cuando existe pie cavo y dedos en garra es necesario calzado con caja anterior amplia (43).
- d)
Dolor: Pueden presentar dolor de origen musculoesquelético o neuropático. Su tratamiento se basa en manejo farmacológico y no farmacológico.
- e)
Manejo nutricional: Es importante mantener un peso adecuado, ya que el sobrepeso y obesidad son factores negativos frente a las deformidades y alteraciones de la marcha.
- f)
Cirugía: Se debe considerar cuando las deformidades interfieren de manera importante en la marcha, cuando son rígidas y no se controlan con tratamiento rehabilitador (43).
Las osteotomías multinivel bien diseñadas y trasposiciones tendinosas pueden corregir deformidades adecuadamente. El tratamiento final de un paciente será la correcta mezcla de cirugías destinadas a corregir las diferentes alteraciones. Por lo tanto, la evidencia para preferir un enfoque sobre otro es escasa y las indicaciones y el momento de la cirugía se basan en la opinión de expertos, siendo la triple artrodesis la cirugía que durante mucho tiempo se ha empleado para tratar las deformidades más severas de los pies, pero se debe evitar en menores de 10 años por sus complicaciones. Se debe reservar este enfoque para casos severos cuando otros enfoques más conservadores no son factibles y la cirugía no puede ser evitada. Osteotomías más limitadas se emplean con frecuencia para corregir deformidades en equino-cavo-varo (38).
La modificación del perfil epidemiológico de los problemas de salud en Chile ha favorecido que las enfermedades crónicas y complejas adquieran mayor relevancia, entre las que se encuentran las ENM. Si bien no existen datos en nuestro país en relación a la real prevalencia de estás, se estima que debería existir al menos 4.500 pacientes con ENM hereditarias (45). El diagnóstico correcto y precoz permite una adecuada información a la familia en relación al consejo genético, pronóstico y tratamiento. Abordaje multidisciplinario a través de protocolos de atención estandarizados para las diferentes patologías, desarrollo de tecnología asistiva, nuevas ayudas técnicas, cirugías oportunas, programas de vacunación especial y ventilación domiciliaria no invasiva entre otros, han permitido cambios importantes en una mayor esperanza y calidad de vida.
El desafío es encontrar una cura para estas enfermedades, avances logrados en terapia génica y celular dan una esperanza para el futuro.
Las autoras declaran no tener conflictos de interés, en relación a este artículo.