



Introducción
El principal objetivo de esta revisión será, en primer lugar, conocer el papel de la mitocondria en el estado bioenergético del condrocito y, en segundo lugar, cómo la disfunción mitocondrial en el condrocito puede interferir con los principales mecanismos patogénicos de la artrosis (OA), al mediar en los mecanismos de reparación de la matriz, calcificación de la matriz, estrés oxidativo y viabilidad del condrocito. Asimismo, evaluaremos potenciales estudios futuros y aplicaciones terapéuticas de la implicación de la mitocondria en la OA.
Mitocondria y estado bioenergético del condrocito
Dado que la síntesis de la matriz extracelular del condrocito y su mineralización están moduladas por el balance entre la generación de adenosintrifosfato (ATP) y su consumo, el mecanismo por el cual el condrocito genera energía constituye un tema primordial1.
El ATP se sintetiza en la célula por 2 mecanismos: a) en el citoplasma mediante las reacciones de oxidación de la glucólisis, y b) en la mitocondria en el ciclo del Krebs y en la vía de la fosforilación oxidativa. En la primera vía, bajo condiciones de baja tensión de oxígeno, la producción de lactato puede regenerar nicotinamida adenina dinucleotido oxidado (NAD+) para permitir la producción glucolítica de ATP. En la segunda vía, en presencia de O2, el piruvato es transportado a la matriz mitocondrial para su posterior oxidación en el ciclo del Krebs, dando lugar a las formas reducidas de las moléculas transportadoras de hidrógeno; es decir, el NADH y el flavín-adenín dinucleótido (FADH2)
que entran en la cadena respiratoria mitocondrial (CRM). Como consecuencia de las funciones redox en la CRM, los protones son bombeados desde la matriz mitocondrial hasta el espacio intermembranoso de la mitocondria, generando un potencial electroquímico de protones. Este gradiente de protones es usado para la síntesis de ATP.
La tensión de O2 en los tejidos es variable, el valor final depende de un gran número de factores. Entre estos factores se incluye el número de células por unidad de volumen y el grado de metabolismo celular, la difusión del O2 a través de la matriz extracelular y el aporte vascular. En este sentido, las células del cartílago articular tienen un aporte sanguíneo muy limitado. Por eso, los condrocitos articulares están expuestos a bajas tensiones de O2, siendo el tejido sinovial, por un lado, y los vasos del hueso, por el otro, la principal fuente de nutrientes2. En este sentido, existe un gradiente en el aporte de O2. Como resultado de este gradiente, los condrocitos de las capas más adyacentes al tejido sinovial están probablemente expuestos a una tensión normal de O2. En contraste, las células de las regiones más profundas del cartílago deben de estar expuestas a una baja tensión de oxígeno. Así, algunos autores sugieren que el reducido grado de respiración aeróbica de las células del cartílago puede ser una respuesta adaptativa a las bajas tensiones de O23. Los resultados de un gran número de experimentos indican que mientras el ciclo del Krebs es activo en el cartílago, un 80% de la glucosa es metabolizado a lactato por glucólisis anaeróbica4. Sin embargo, la actividad mitocondrial de los condrocitos humanos normales en cultivo produce ATP, y la actividad de la CRM muestra una actividad enzimática muy similar a la de otras células mesenquimales (tabla 1)5.

Interacción de la mitocondria del condrocito con los mecanismos patogénicos de la artrosis
Las mitocondrias de los condrocitos aumentan en número y tamaño en la zona superficial y media cuando la OA comienza a establecerse. Asimismo, el análisis de la actividad del trasporte de electrones en condrocitos OA refleja una disminución significativa de la actividad de los complejos II y III de la CRM en relación con los condrocitos normales (tabla 1)5. Esta disfunción en los complejos II y III compromete la transferencia de electrones y este defecto podría ser solventado por una sobrecarga en la transferencia de electrones vía complejo I, una vía con un pequeño consumo de oxígeno. Sin embargo, la disminución en la eficacia en el trasporte de electrones vía complejo II no aumenta la actividad del complejo I. Por otro lado, la masa mitocondrial está incrementada en el cartílago OA, como se demuestra por un aumento significativo de la actividad citrato sintasa (CS). En este sentido, el incremento de la masa mitocondrial puede ser un mecanismo de los condrocitos OA para compensar la deficiencia en la trasferencia de electrones vía complejos II y III y como resultado de la baja producción de ATP por la mitocondria5.
Una generación insuficiente de ATP mitocondrial puede ser uno de los factores responsables del fallo metabólico de la OA. En este sentido, la inhibición de la síntesis de ATP con un inhibidor del complejo II, como es la antimicina, y un inhibidor del complejo V, la oligomicina, a dosis subletales, causa una marcada inhibición de la síntesis de colágeno y proteoglicanos y un incremento en la síntesis de la matriz calcificada, situación que reproduce fenómenos característicos de la enfermedad OA6. Los condrocitos OA demuestran una disminución en la actividad de síntesis de la matriz y en la respuesta proliferativa al factor de crecimiento insulínico (IGF-1). Asimismo, en un trabajo reciente, se demuestra que la capacidad del factor transformador del crecimiento (TGF-ß) de aumentar los valores intracelulares de ATP en condrocitos en cultivo está debilitado por la supresión de la fosforilación oxidativa6.
Varios estudios demuestran que el cartílago OA presenta niveles elevados de apoptosis in situ. Una celularidad reducida también desarrolla cartílagos OA. En este contexto, la mitocondria desempeña un papel principal en la regulación de la apoptosis7. Interesante es señalar que las mitocondrias de los condrocitos OA presentan disminuido el potencial de membrana mitocondrial (*m)5. En este sentido, el colapso del *m está asociado con la hinchazón de la mitocondria, la ruptura de la membrana mitocondrial externa y la liberación de factores proapoptóticos como el citocromo c, Apaf-1 y procaspasas del espacio intermembranoso. Bcl-2 es uno de los componentes de la familia de proteínas antiapoptóticas que se localiza en la membrana externa de la mitocondria y que previene la permeabilización de la mitocondria y la liberación del citocromo c. Es interesante destacar que el cartílago OA muestra una expresión muy marcada de Bcl-2. La caspasa-3 es una de las caspasas ejecutoras más importantes que completan el proceso de apoptosis después de la activación de la caspasa-9. El citocromo c liberado desde la mitocondria se une a Apaf-1 y este complejo activa la procaspasa-9. Un incremento en la expresión de caspasa-3 se ha confirmado en cartílago articular humano OA y en modelos experimentales de OA. Esta expresión también se correlaciona con el grado de apoptosis y con las lesiones OA7.
La mitocondria puede participar en el desarrollo de la OA al mediar los efectos citotóxicos de la inflamación. Así, dependiendo del tipo celular, la mitocondria puede ser un efector de la actividad citotóxica, como consecuencia de una disminución del transporte de electrones mitocondrial y de la generación de especies reactivas de O2 mitocondriales; o bien, disminuir la citotoxicidad al inducir la expresión de superóxido dismutasa. Actualmente, la teoría más extendida es que la mitocondria puede actuar amplificando la señal apoptótica inducida por el factor de necrosis tumoral alfa (TNF-*) a través de la escisión de Bid que promueve la liberación de citocromo c por la mitocondria1. En relación con la quimiocina IL-8, que está regulada en los condrocitos OA in situ, se ha demostrado que es inducida por la hipoxia en hepatocitos, lo que refleja las alteraciones en el estado redox mitocondrial.
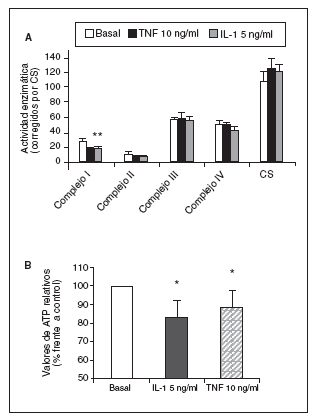
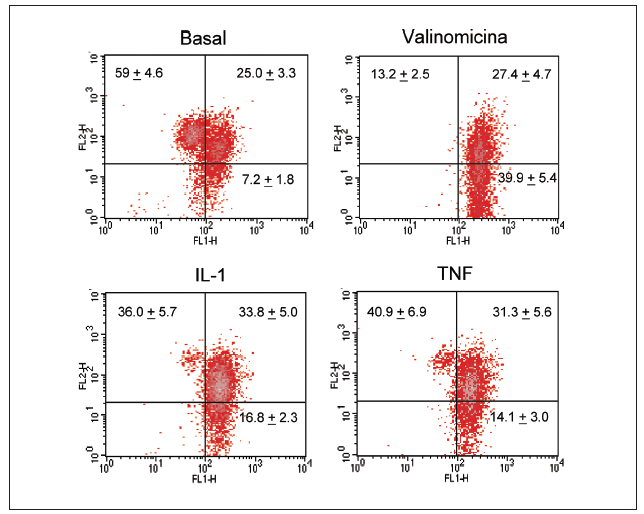
Significativamente, la interleucina (IL)-1ß y el TNF-* inducen en condrocitos la disminución de la actividad del complejo I, acompañada de la disminución en los valores de ATP, disminución del *m (figs. 1 y 2) y modulación de la familia de proteínas Bcl-2 y de caspasas8. Se especula que la disfunción de la CRM bajo ciertas condiciones crónicas de inflamación, y la presencia de la generación de óxido nítrico (NO) incrementada en el cartílago OA amplifica la generación de especies reactivas de O2 (ROS) por los condrocitos con consecuencias funcionales para el condrocito. En este contexto, se ha observado que la producción de ROS disminuye la cantidad de transcritos mitocondriales en condrocitos articulares de conejo en cultivo. La generación aumentada de ROS puede, en sí misma, modular las señales de transducción en parte a través de los efectos de los factores de trascripción proteína activadora-1 (AP-1) y factor nuclear-kappaB (NF-kB), que pueden promover la expresión de ciertas enzimas degradadoras de la matriz; estos genes incluyen las metaloproteasas (MMP), que como se conoce están sobreexpuestas en el cartílago OA1.
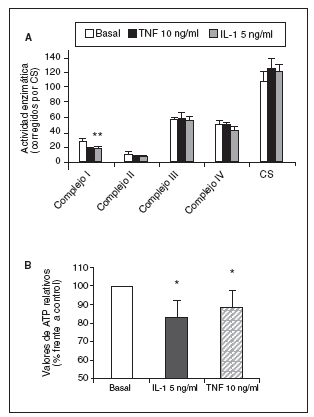
Figura 1. Modulación de la actividad de los complejos de la cadena respiratoria mitocondrial (CRM) (A) y valores de adenosintrifosfato (ATP) (B) por el factor de necrosis tumoral alfa (TNF-*) y la interleucina-1-beta (IL-1ß) en condrocitos humanos en cultivo. CS: citrato sintasa. *p < 0,05.
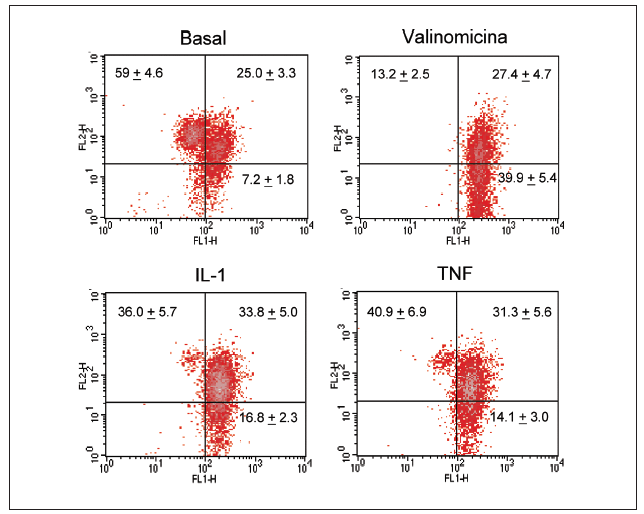
Figura 2. Despolarización del potencial de membrana mitocondrial por el TNF-* y la IL-1 en condrocitos humanos en cultivo. Los condrocitos humanos fueron incubados durante 12 h en medio o en presencia de TNF-* o IL-1ß antes de que las células fueran teñidas con DePhefiser y analizadas por citometría de flujo, para cuantificar el potencial de membrana mitocondrial. Como control positivo se empleó la valinomicina (1 µM). Un plot de densidades representativo se muestra para cada condición. El fluoróforo nos indica si existe despolarización de la membrana según 2 tipos de fluorescencia: fluorescencia verde (FL-1) y fluorescencia roja (FL-2), ya que el fluoróforo tiene la propiedad de agregarse y penetrar en el espacio intermembranoso emitiendo fluorescencia roja cuando el potencial de membrana mitocondrial no está alterado. Si el potencial disminuye, el fluoróforo no accede al espacio intermembranoso permaneciendo en el citosol en forma monomérica. Los números representan el porcentaje de cada población.
En la patogénesis de la OA se incluye la elaboración de grandes cantidades de óxido nítrico (NO) como consecuencia de la sobreexpresión de la NO sintasa inducible (iNOS), cuya expresión se incrementa por IL-1ß, TNF-* y otros factores9. El NO ha demostrado ser capaz de disminuir la producción de energía mitocondrial; además este efecto está aumentado con bajas tensiones de O2. Una posible explicación para el hecho de que el NO induzca apoptosis es la interacción adicional con ROS. Un incremento en los valores intracelulares de las ROS es una condición demostrada bajo la cual el NO puede ser citotóxico vía formación de otras especies reactivas de nitrógeno (RNS)10.
Mitocondria y mineralización del cartílago en la osteoartritis
Algunos estudios sugieren que las mitocondrias del condrocito están especializadas en el transporte de Ca2+ y que son importantes en la calcificación de la matriz extracelular. En este sentido, la condrocalcinosis (calcificación patológica de la matriz del cartílago), que se manifiesta como depósito de cristales de pirofosfato cálcico dihidratado (CPPD) y/o hidroxiapatita (HA), es un hecho común en la OA1. Estos cristales son capaces de inducir directamente citotoxicidad en los condrocitos, así como inflamación sinovial y también pueden promover episodios sintomáticos y empeoramiento de la articulación OA.
La calcificación de la matriz en condrocitos puede ser estimulada por el NO y el peroxinitrito. Además, la calcificación de la matriz del cartílago se estimula de forma directa con la apoptosis, y la apoptosis del condrocito se colocaliza con la matriz del cartílago calcificada en el cartílago OA. La calcificación de la matriz del cartílago también es modulada por el metabolismo del ATP11. Específicamente, el ATP es la principal fuente del pirofosfato inorgánico (PPi) liberado por los condrocitos. El PPi potencialmente suprime la propagación de los cristales de HA, y los valores fisiológicos de PPi extracelular mantenidos por los condrocitos por un mecanismo dependiente de la generación de ATP mitocondrial son relativamente altos comparados con otros tejidos, y ello previene al cartílago articular normal de la calcificación. Así, un depósito patológico de cristales de HA y CPPD es un hecho frecuente en OA, especialmente en estados avanzados de la enfermedad. Es importante destacar que el inicio del depósito de cristales HA alrededor de los condrocitos en el cartílago hipertrófico, coincide con un cambio brusco en el estado metabólico y redox de los condrocitos, en asociación con la pérdida local de respiración celular11. En este sentido, la supresión directa de la respiración promueve la mineralización de las vesículas de la matriz. Siendo así, la actividad de la CRM puede parcialmente regular el depósito diferencial de HA y CPPD. El NO puede modular la mineralización patológica del cartílago a través de la interacción con la CRM.
Estudios futuros de la implicación de la mitocondria en la artrosis
Una aproximación para conocer el papel de la mitocondria en la OA es determinar los efectos de la inhibición de la CRM y compararlos con los encontrados en la enfermedad OA. La inhibición de la CRM con antimicina (inhibidor del complejo III de la CRM) previene la capacidad normal del TGF-ß de incrementar la secreción de Pi, empeorando el deposito patológico de cristales de HA11. La inhibición de la generación de ATP mitocondrial con antimicina y oligomicina, inhibidores de la CRM, está asociada con una disminución de la síntesis de colágeno y proteoglicanos. Debido a que la interacción con las proteínas de la matriz pericelular es un factor central en la regulación de apoptosis, la desregulación de la actividad de la CRM de los condrocitos, así como la generación de ROS por la disturbación del transporte de electrones mitocondrial, presenta la capacidad de contribuir al incremento de apoptosis observado in situ en los condrocitos OA. Se ha descrito que el NO puede directamente inhibir el complejo IV debido a su unión reversible con la citocromo c oxidasa en competición con el O2. En los condrocitos la inhibición del complejo IV con azida sódica modifica tanto el potencial de membrana como la supervivencia de las células. La inhibición del complejo I con rotenona en condrocitos humanos normales incrementa la expresión y la síntesis de Bcl-2 y COX-2, con un efecto similar al producido por IL-1ß y TNF-* en condrocitos humanos.
Por otro lado, algunos estudios preliminares soportan la hipótesis de que los defectos sobre la actividad mitocondrial en los condrocitos son irreversibles. Una posible explicación para este fenómeno puede deberse a que las ROS inducen mutaciones somáticas en los genes mitocondriales (ADN geonómico y mitocondrial). Por esta razón, los estudios genómicos y proteómicos de la disfunción mitocondrial en condrocitos OA pueden ser la base para proporcionar en el futuro un conocimiento más detallado de la relación entre mitocondria y enfermedad OA.
Asimismo, dado que la OA no sólo afecta al cartílago articular, sino que involucra a toda la estructura articular, incluyendo el hueso subcondral, ligamentos, cápsula, membrana sinovial y músculos periarticulares, en aproximaciones futuras se debería analizar el papel de la mitocondria en la patología de estos tejidos.
Finalmente, una aproximación obligada a desarrollar en los próximos años es demostrar la capacidad de algunos compuestos para preservar la función mitocondrial.
Aplicaciones terapéuticas de la implicación de la mitocondria en la artrosis
El tratamiento médico de la OA inicialmente se ha dirigido al control de los síntomas diarios de dolor e inflamación, usando antiinflamatorios no esteroideos (AINE) y una gran variedad de otros antiinflamatorios y agentes analgésicos. Todos estos tratamientos de la OA nos permiten controlar el dolor, pero han sido altamente ineficaces en prevenir el progresivo deterioro de las articulaciones afectadas. En este sentido, intentando resolver este punto, se han diseñado diversas estrategias en el tratamiento de la OA que incluye desde inhibidores de iNOS, inhibidores de la síntesis de IL-1ß o unión a su receptor, el sulfato de glucosamina, el candroitín sulfato y el ácido hialurónico intraarticular. Todos estos tratamientos tienen la capacidad de actuar, al menos en parte, en la función mitocondrial vía efectos antioxidantes y preservación de la fosforilación oxidativa.
Las limitaciones potenciales de una aproximación terapéutica común al desarrollo de otros fármacos es consecuencia de que el fármaco debe de actuar en una organela específica del condrocito de cartílago articular no vascularizado.
En resumen, el papel de la mitocondria en OA se está analizando actualmente, y son varias las evidencias que sugieren una posible relación entre la disfunción de la mitocondria en los condrocitos y la degradación del cartílago.
Agradecimientos
La doctora López-Armada es investigadora del Programa Ramón y Cajal (Ministerio de Educación).