








Las proteínas mitocondriales pueden ser modificadas por reacciones de glicación inducidas por compuestos dicarbonilo procedentes del metabolismo tales como el glioxal y el metilglioxal. Estas modificaciones provocan cambios estructurales y funcionales en las proteínas implicadas. La modificación del proteoma mitocondrial por estos compuestos dicarbonilo puede inducir disfunción mitocondrial y acentuar un estado de estrés oxidativo. Así, estas modificaciones químicas podrían jugar un papel clave en el proceso fisiológico de envejecimiento y patologías asociadas a la edad, donde se han evidenciado tanto defectos de la actividad mitocondrial como incrementos de compuestos dicarbonilo. Identificar las proteínas mitocondriales especificamente modificadas, aseverar los cambios funcionales derivados y sus implicaciones constituyen lineas de investigación que tendrán su desarrollo en los próximos años.
Mitochondrial proteins can be modified by glycation reactions from endogenous dicarbonyl compounds such as physiologically generated methylglyoxal and glyoxal. This modification could cause structural and functional changes in the proteins Consequently, dicarbonyl attack of the mitochondrial proteome may be an event leading to mitochondrial dysfunction and thus, to oxidative stress. These protein chemical modifications can play an important role in the physiological aging process and age-associated diseases, where both mitochondrial deffects and increased dicarbonyl concentrations have been found. Future research should address the functional changes in mitochondrial proteins that are the targets for dicarbonyl glycation.
Los estudios experimentales basados en mutaciones que afectan la longevidad de organismos diversos tales como levaduras, nemátodos (Caenorhabditis elegans), moscas (Drosophila melanogaster) y ratones, demuestran que la generación de especies reactivas derivadas del oxígeno (ROS) y, por extensión, el estrés oxidativo, juega un papel causal clave en el proceso de envejecimiento1.
En este contexto, en un estudio reciente2 se describía un nuevo mecanismo capaz de regular la producción mitocondrial de ROS y la longevidad en C. elegans: la progresiva modificación de proteínas mitocondriales por metilglioxal (MG), un metabolito dicarbonilo derivado de glicólisis. Los autores de dicho estudio constataron que la sobreexpresión del gen para la glioxalasa 1 (Glo1) en el nemátodo C. elegans inducía un aumento de la longevidad. Por el contrario, el silenciamiento de Glo1 reducía dicha longevidad. Glo1 es otro ejemplo de un «vitagene», un gen que cuando se manipula para cambiar su expresión induce un cambio en la longevidad3. Glo1 es una enzima dependiente de glutatión (GSH) que cataliza el metabolismo de compuestos dicarbonilo reactivos tales como MG y glioxal4 previniendo, de este modo, la modificación química de proteínas mediada por dichos compuestos (glicación) (fig. 1). Observaron, asímismo, que las proteínas mitocondriales eran dianas prioritarias de la glicación por dicarbonilos constatándose, además, que un aumento en la glicación de proteínas mitocondriales se asociaba con un aumento de la formación de ROS y un incremento de la lesión del proteoma por procesos oxidativos y nitrosativos. En dicho estudio también se demostró que el proceso de envejecimiento fisiológico de C. elegans inducía un declive de la expresión de Glo1 y un aumento de la generación de ROS de origen mitocondrial. La sobreexpresión de Glo1 en C. elegans disminuía la glicación por dicarbonilos de las proteínas mitocondriales, así como la formación de ROS, los marcadores de glicación por dicarbonilos del proteoma y también los marcadores de lesión oxidativa y nitrosativa (metionina sulfóxido y 3-nitrotirosina, respectivamente), con un aumento concomitante de la longevidad. Esto indicaba, por primera vez, que la glicación por dicarbonilos podría ser una lesión crítica del proteoma mitocondrial que podría disparar la producción de ROS y la lesión oxidativa durante el proceso de envejecimiento.
 Formación de productos de glicación inicial y AGE a partir de glucosa y productos intermediarios glicolíticos y de peroxidación lipídica. B) Compuestos dicarbonilo reactivos generados fisiológicamente.")
Principales rutas de formación de productos AGE en los sitemas fisiológicos y sus principales metabolitos dicarbonilo precursores. A) Formación de productos de glicación inicial y AGE a partir de glucosa y productos intermediarios glicolíticos y de peroxidación lipídica. B) Compuestos dicarbonilo reactivos generados fisiológicamente.
La glicación es una de las principales causas de modificación química (lesión) espontánea de proteínas celulares y extracelulares en los sistemas fisiológicos, afectando a un 0,1–0,2% del total de residuos de lisina y arginina5,6. La glicación por glucosa y otros monosacáridos está dirigida principalmente a grupos amino de residuos de lisina y aminoácidos N-terminales de proteínas, así como residuos de arginina y cisteína7,8.
Para unas condiciones de reactividad dada (temperatura, concentración de sustratos y accesibilidad a los grupos químicos reactivos de los aminoácidos, entre otros), uno de los principales factores que regula el estado basal de lesión es la vida media proteica, o en otras palabras, su tasa de recambio. Para algunas proteínas con un recambio limitado o nulo, tales como las del cristalino, el grado de extensión de la glicación proteica puede llegar a ser hasta 10 veces superior9. La mayoría de las proteínas mitocondriales son, sin embargo, de vida corta, con unas tasas de recambio que van de 10–30min a 3–5 días10. El «control de calidad» del proteoma mitocondrial implica a proteasas mitocondriales, que conducen a la liberación de péptidos desde la mitocondria11, ubicuitilación y proteolisis de la membrana externa, fisión y fusión mitocondrial, y finalmente autofagia de las mitocondrias lesionadas (mitofagia)12.
Los productos o compuestos derivados de la fase inicial de glicación se forman en reacciones espontáneas no enzimáticas de monosacáridos con proteínas (fig. 1A). La glucosa entraría en la mitocondria a través del transportador de membrana GLUT1 (transportador de glucosa 1), que también transporta vitamina C al interior de la mitocondria.13 Dada la baja reactividad de la glucosa per se, los agentes glicantes más importantes a considerar para la lesión por glicación del proteoma mitocondrial son compuestos dicarbonilo reactivos tales como glioxal, MG y 3-deoxiglucosona (fig. 1B).
Los compuestos dicarbonilo se forman endógenamente por peroxidación lipídica y degradación de intermediarios glicolíticos y proteínas glicadas. Estos compuestos dicarbonilo son potentes agentes glicantes, entre 200–50.000 veces más reactivos que la glucosa. No obstante, las concentraciones fisiológicas de dicarbonilos típicamente son del orden de 10.000–50.000 veces inferior que la de glucosa. Aún así, los compuestos dicarbonilos continúan siendo, en los sitemas fisiológicos, los precursores más importantes y determinantes de la formación de los denominados productos de glicación avanzada (AGE).
Estadios iniciales de los productos de glicación y AGELos aductos (entendido como compuestos químicos originados por combinación directa de dos especies químicas que mantienen en aquel su respectiva ordenación atómica) de glicación originados por glucosa y otros monosacáridos se denominan aductos de glicación de estadios iniciales (fig. 1). El producto de glicación inicial predominante es la Nε-fructosil-lisina (FL). La base de Schiff y aductos FL pueden degradarse lentamente en posteriores reacciones avanzadas dando lugar a la formación de numerosos y diferentes aductos de glicación. A estos aductos, de carácter final e irreversible, se les conoce colectivamente como AGE. Sin embargo, los compuestos dicarbonilo (derivados del metabolismo) reaccionan directamente con las proteínas para formar también AGE. Los estudios mediante espectrometría de masas demuestran que entre los AGE cuantitativamente importantes se cuentan las hidroimidazolonas derivadas de residuos de arginina modificados por glioxal, MG y 3-deoxiglucosona: G-H1 (Nδ-[5-hidro-4-imidazolon-2-il]ornitina), MG-H1 (Nδ-[5-hidro-5-metil-4-imidazolon-2-il]-ornitina) y 3-deoxiglucosona-H (Nδ-[5-hidro-5-2,3,4-trihidroxibutil-4-imidazolon-2-il]ornitina e isómeros estructuralmente relacionados). Otros AGE ampliamente estudiados e importantes comprenden CML (Nε-carboximetil-lisina) y CEL (Nε-carboxietil-lisina), y los compuestos entrecruzados bis-lisil GOLD (derivado de glioxal) y MOLD (derivado de MG), y los compuestos AGE que también generan entrecruzamientos pentosidina y glucosapana6,13–18. Otros compuestos AGE y derivados relacionados de importancia emergente son CMC (S-carboximetil-cisteína)19, CMA (Nε-carboximetilarginina)20 y ornitina21, este último generado como producto de degradación de hidroimidazolonas (fig. 2).
Defensas enzimáticas contra la glicación
Existe un conjunto diverso de enzimas que previenen o reparan la glicación de proteínas en los sistemas fisiológicos22,23. Este mecanismo de defensa involucra actividades enzimáticas que evitan la formación de los aductos de glicación y también aquellas que «reparan» los lugares de glicación inicial. Entre las primeras se enumerarían Glo1, aldo-ceto reductasas y aldehido deshidrogenasa cuyas actividades mantienen dentro de los límites fisiológicos las concentraciones de dicarbonilos reactivos que se comportan como agentes glicantes24,25; y entre las segundas se contaría con amadoriasas y fructosamina 3-fosfoquinasas, que catalizan la eliminación de residuos FL y aductos libres (fig. 3)26–28.

La detección de los aductos de glicación en proteínas, aminofosfolípidos y ácidos nucleicos18, y el reducido grado de modificación existente en condiciones fisiológicas, nos indican que la defensa enzimática contra la glicación limita el impacto de la lesión a las macromoléculas biológicas, pero que se trata de una defensa imperfecta. Así, en algunos estados patológicos (p. ej.: diabetes, insuficiencia renal crónica y enfermedades neurodegenerativas, entre muchas otras), estos mecanismos de defensa son desbordados, y las concentraciones de los aductos de glicación aumentan. Cabe postular que la progresiva pérdida de expresión de enzimas de defensa contra la glicación podría ser clave en el incremento de glicación de proteínas detectado durante el proceso de envejecimiento.
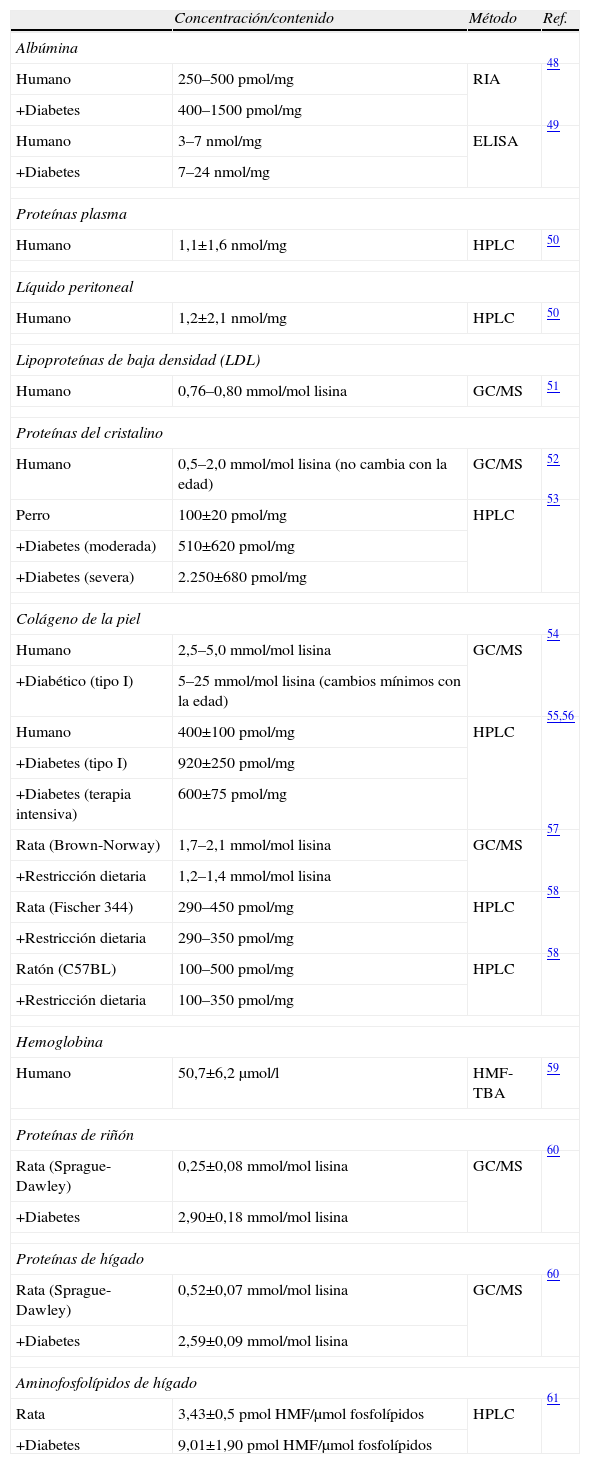
In vivo, el contenido en productos AGE difiere dependiendo de las características de la/s proteína/s (composición en aminoácidos, estructura tridimensional, tasa de recambio, etc.), su localización tisular y el tipo de AGE. Así, se han descrito niveles relativamente elevados de FL (0,1–10mmol/mol de aminoácido modificado) y MG-H1 (0,1–15mmol/mol aminoácido modificado), un contenido menor de CML, CEL y CMC (0,05–6mmol/mol lisina) y cantidades traza de GOLD y MOLD (0,001–0,002mmol/mol lisina). Las concentraciones más elevadas de FL e hidroimidazolonas han sido descritas en proteínas de cristalino y colágeno de piel de personas de edad avanzada. El contenido de los demás compuestos AGE, sin llegar a los niveles de los anteriores, se han descrito que aumentan con la edad en proteínas de cristalino, matriz extracelular y proteínas de diversos tejidos y especies (tablas 1–3)18.
Niveles de Nε-fructosil-lisina en diferentes proteínas y tejidos de mamífero y en diferentes condiciones fisiológicas y patológicas¿
| Concentración/contenido | Método | Ref. | |
| Albúmina | |||
| Humano | 250–500pmol/mg | RIA | 48 |
| +Diabetes | 400–1500pmol/mg | ||
| Humano | 3–7nmol/mg | ELISA | 49 |
| +Diabetes | 7–24nmol/mg | ||
| Proteínas plasma | |||
| Humano | 1,1±1,6nmol/mg | HPLC | 50 |
| Líquido peritoneal | |||
| Humano | 1,2±2,1nmol/mg | HPLC | 50 |
| Lipoproteínas de baja densidad (LDL) | |||
| Humano | 0,76–0,80mmol/mol lisina | GC/MS | 51 |
| Proteínas del cristalino | |||
| Humano | 0,5–2,0mmol/mol lisina (no cambia con la edad) | GC/MS | 52 |
| Perro | 100±20pmol/mg | HPLC | 53 |
| +Diabetes (moderada) | 510±620pmol/mg | ||
| +Diabetes (severa) | 2.250±680pmol/mg | ||
| Colágeno de la piel | |||
| Humano | 2,5–5,0mmol/mol lisina | GC/MS | 54 |
| +Diabético (tipo I) | 5–25mmol/mol lisina (cambios mínimos con la edad) | ||
| Humano | 400±100pmol/mg | HPLC | 55,56 |
| +Diabetes (tipo I) | 920±250pmol/mg | ||
| +Diabetes (terapia intensiva) | 600±75pmol/mg | ||
| Rata (Brown-Norway) | 1,7–2,1mmol/mol lisina | GC/MS | 57 |
| +Restricción dietaria | 1,2–1,4mmol/mol lisina | ||
| Rata (Fischer 344) | 290–450pmol/mg | HPLC | 58 |
| +Restricción dietaria | 290–350pmol/mg | ||
| Ratón (C57BL) | 100–500pmol/mg | HPLC | 58 |
| +Restricción dietaria | 100–350pmol/mg | ||
| Hemoglobina | |||
| Humano | 50,7±6,2μmol/l | HMF-TBA | 59 |
| Proteínas de riñón | |||
| Rata (Sprague-Dawley) | 0,25±0,08mmol/mol lisina | GC/MS | 60 |
| +Diabetes | 2,90±0,18mmol/mol lisina | ||
| Proteínas de hígado | |||
| Rata (Sprague-Dawley) | 0,52±0,07mmol/mol lisina | GC/MS | 60 |
| +Diabetes | 2,59±0,09mmol/mol lisina | ||
| Aminofosfolípidos de hígado | |||
| Rata | 3,43±0,5pmol HMF/μmol fosfolípidos | HPLC | 61 |
| +Diabetes | 9,01±1,90pmol HMF/μmol fosfolípidos | ||
ELISA: enzyme linked inmunoabsorvent assay; GC/MS: gas chromatography/mass spectrometry; HMF-TBA: hydroxymethylfurfural-thiobarbituric acid method; HPLC: high performance liquid chromatography; RIA: radioimmnuoassay.
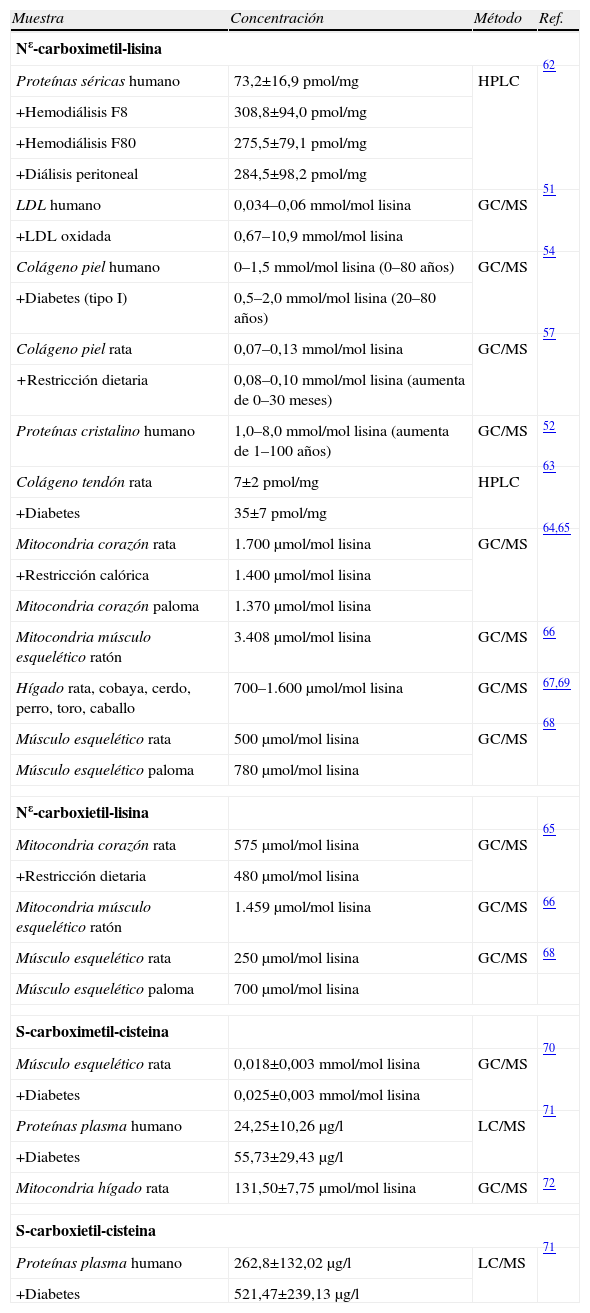
Niveles de productos AGE en diferentes proteínas y tejidos de mamífero y en diferentes condiciones fisiológicas y patológicas¿
| Muestra | Concentración | Método | Ref. |
| Nε-carboximetil-lisina | |||
| Proteínas séricas humano | 73,2±16,9pmol/mg | HPLC | 62 |
| +Hemodiálisis F8 | 308,8±94,0pmol/mg | ||
| +Hemodiálisis F80 | 275,5±79,1pmol/mg | ||
| +Diálisis peritoneal | 284,5±98,2pmol/mg | ||
| LDL humano | 0,034–0,06mmol/mol lisina | GC/MS | 51 |
| +LDL oxidada | 0,67–10,9mmol/mol lisina | ||
| Colágeno piel humano | 0–1,5mmol/mol lisina (0–80 años) | GC/MS | 54 |
| +Diabetes (tipo I) | 0,5–2,0mmol/mol lisina (20–80 años) | ||
| Colágeno piel rata | 0,07–0,13mmol/mol lisina | GC/MS | 57 |
| +Restricción dietaria | 0,08–0,10mmol/mol lisina (aumenta de 0–30 meses) | ||
| Proteínas cristalino humano | 1,0–8,0mmol/mol lisina (aumenta de 1–100 años) | GC/MS | 52 |
| Colágeno tendón rata | 7±2pmol/mg | HPLC | 63 |
| +Diabetes | 35±7pmol/mg | ||
| Mitocondria corazón rata | 1.700μmol/mol lisina | GC/MS | 64,65 |
| +Restricción calórica | 1.400μmol/mol lisina | ||
| Mitocondria corazón paloma | 1.370μmol/mol lisina | ||
| Mitocondria músculo esquelético ratón | 3.408μmol/mol lisina | GC/MS | 66 |
| Hígado rata, cobaya, cerdo, perro, toro, caballo | 700–1.600μmol/mol lisina | GC/MS | 67,69 |
| Músculo esquelético rata | 500μmol/mol lisina | GC/MS | 68 |
| Músculo esquelético paloma | 780μmol/mol lisina | ||
| Nε-carboxietil-lisina | |||
| Mitocondria corazón rata | 575μmol/mol lisina | GC/MS | 65 |
| +Restricción dietaria | 480μmol/mol lisina | ||
| Mitocondria músculo esquelético ratón | 1.459μmol/mol lisina | GC/MS | 66 |
| Músculo esquelético rata | 250μmol/mol lisina | GC/MS | 68 |
| Músculo esquelético paloma | 700μmol/mol lisina | ||
| S-carboximetil-cisteina | |||
| Músculo esquelético rata | 0,018±0,003mmol/mol lisina | GC/MS | 70 |
| +Diabetes | 0,025±0,003mmol/mol lisina | ||
| Proteínas plasma humano | 24,25±10,26μg/l | LC/MS | 71 |
| +Diabetes | 55,73±29,43μg/l | ||
| Mitocondria hígado rata | 131,50±7,75μmol/mol lisina | GC/MS | 72 |
| S-carboxietil-cisteina | |||
| Proteínas plasma humano | 262,8±132,02μg/l | LC/MS | 71 |
| +Diabetes | 521,47±239,13μg/l | ||
GC/MS: gas chromatography/mass spectrometry; HPLC: high performance liquid chromatography; LC/MS: liquid chromatography/mass spectrometry.
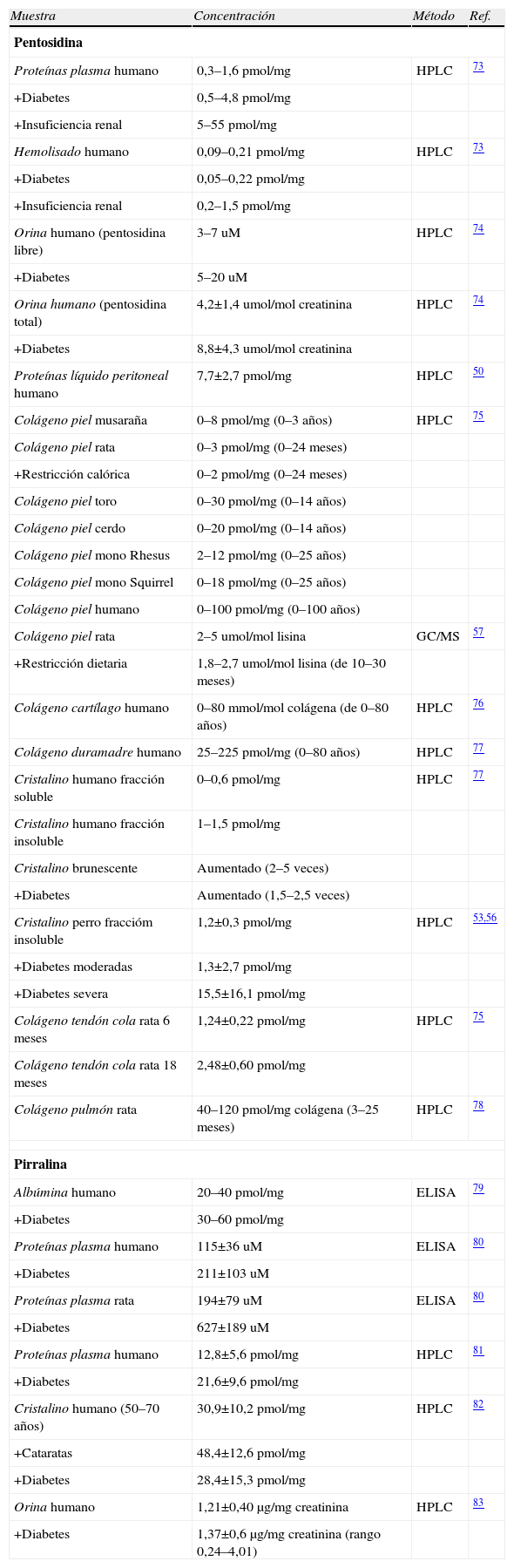
Niveles de los productos AGE pentosidina y pirralina en diferentes proteínas y tejidos de mamífero y en diferentes condiciones fisiológicas y patológicas¿
| Muestra | Concentración | Método | Ref. |
| Pentosidina | |||
| Proteínas plasma humano | 0,3–1,6pmol/mg | HPLC | 73 |
| +Diabetes | 0,5–4,8pmol/mg | ||
| +Insuficiencia renal | 5–55pmol/mg | ||
| Hemolisado humano | 0,09–0,21pmol/mg | HPLC | 73 |
| +Diabetes | 0,05–0,22pmol/mg | ||
| +Insuficiencia renal | 0,2–1,5pmol/mg | ||
| Orina humano (pentosidina libre) | 3–7uM | HPLC | 74 |
| +Diabetes | 5–20uM | ||
| Orina humano (pentosidina total) | 4,2±1,4umol/mol creatinina | HPLC | 74 |
| +Diabetes | 8,8±4,3umol/mol creatinina | ||
| Proteínas líquido peritoneal humano | 7,7±2,7pmol/mg | HPLC | 50 |
| Colágeno piel musaraña | 0–8pmol/mg (0–3 años) | HPLC | 75 |
| Colágeno piel rata | 0–3pmol/mg (0–24 meses) | ||
| +Restricción calórica | 0–2pmol/mg (0–24 meses) | ||
| Colágeno piel toro | 0–30pmol/mg (0–14 años) | ||
| Colágeno piel cerdo | 0–20pmol/mg (0–14 años) | ||
| Colágeno piel mono Rhesus | 2–12pmol/mg (0–25 años) | ||
| Colágeno piel mono Squirrel | 0–18pmol/mg (0–25 años) | ||
| Colágeno piel humano | 0–100pmol/mg (0–100 años) | ||
| Colágeno piel rata | 2–5umol/mol lisina | GC/MS | 57 |
| +Restricción dietaria | 1,8–2,7umol/mol lisina (de 10–30 meses) | ||
| Colágeno cartílago humano | 0–80mmol/mol colágena (de 0–80 años) | HPLC | 76 |
| Colágeno duramadre humano | 25–225pmol/mg (0–80 años) | HPLC | 77 |
| Cristalino humano fracción soluble | 0–0,6pmol/mg | HPLC | 77 |
| Cristalino humano fracción insoluble | 1–1,5pmol/mg | ||
| Cristalino brunescente | Aumentado (2–5 veces) | ||
| +Diabetes | Aumentado (1,5–2,5 veces) | ||
| Cristalino perro fraccióm insoluble | 1,2±0,3pmol/mg | HPLC | 53,56 |
| +Diabetes moderadas | 1,3±2,7pmol/mg | ||
| +Diabetes severa | 15,5±16,1pmol/mg | ||
| Colágeno tendón cola rata 6 meses | 1,24±0,22pmol/mg | HPLC | 75 |
| Colágeno tendón cola rata 18 meses | 2,48±0,60pmol/mg | ||
| Colágeno pulmón rata | 40–120pmol/mg colágena (3–25 meses) | HPLC | 78 |
| Pirralina | |||
| Albúmina humano | 20–40pmol/mg | ELISA | 79 |
| +Diabetes | 30–60 pmol/mg | ||
| Proteínas plasma humano | 115±36uM | ELISA | 80 |
| +Diabetes | 211±103uM | ||
| Proteínas plasma rata | 194±79uM | ELISA | 80 |
| +Diabetes | 627±189uM | ||
| Proteínas plasma humano | 12,8±5,6pmol/mg | HPLC | 81 |
| +Diabetes | 21,6±9,6pmol/mg | ||
| Cristalino humano (50–70 años) | 30,9±10,2pmol/mg | HPLC | 82 |
| +Cataratas | 48,4±12,6pmol/mg | ||
| +Diabetes | 28,4±15,3pmol/mg | ||
| Orina humano | 1,21±0,40μg/mg creatinina | HPLC | 83 |
| +Diabetes | 1,37±0,6μg/mg creatinina (rango 0,24–4,01) | ||
ELISA: enzyme linked inmunoabsorvent assay; GC/MS: gas chromatography/mass spectrometry; HPLC: high performance liquid chromatography.
La dilucidación de la importancia fisiológica de la glicación de proteínas está actualmente siendo motivo de una intensa actividad científica. No obstante, se han descrito algunos efectos particularmente perjudiciales que afectan a la estructura y función proteica (tabla 4). Así, cabe destacar los efectos nocivos originados por la presencia de entrecruzamientos intra e interproteína que confieren resistencia a la proteolisis; así como los efectos perjudiciales derivados de la modificación de aminoácidos localizados específicamente en aquellos lugares que determinan interacciones proteína-proteína, sustrato-enzima o incluso proteína-ADN, este último especialmente relevante para factores de transcripción.
Efectos de la modificación proteica por productos AGE sobre las propiedades físico-químicas y biológicas
|
|
AGE: productos de glicación avanzada.
Especial mención requiere la modificación potencial de los aminoácidos cisteína y metionina. Cisteína y metionina son los únicos aminoácidos que contienen grupos funcionales (grupos sulfidrilo) que pueden oxidarse de forma reversible bajo diferentes condiciones fisiológicas. Estos grupos químicos se describen como «interruptores» porque su oxidación reversible proporciona un medio de control de la estructura y función proteica. Los grupos sulfidrilo (-SH) de cisteína y metionina pueden sufrir numerosas modificaciones oxidativas, siendo la más estudiada la oxidación reversible de tiol a disulfuro. Cambios en el estado de oxidación/reducción (redox) de tiol/disulfuro afectan la conformación de la proteína, la actividad enzimática, la actividad de transportadores, la unión de ligando a su receptor, las interacciones proteína-proteína, las interacciones proteína-ADN, el tráfico de proteínas y la degradación de las mismas29. Dado que estas propiedades estructurales y funcionales estan reguladas por reacciones de carácter reversible, cabe plantearse qué sucedería si dichas modificaciones fueran irreversibles, como es el caso en la formación del compuesto de glicación CMC.
Glicación de proteínas mitocondrialesLa modificación de proteínas por productos AGE a nivel mitocondrial ha sido detectada por inmunoensayo y por GC-MS. El inmunoensayo de AGE adolece del problema de caracterización incompleta del epítopo reconocido por los anticuerpos. Así, por ejemplo, el anticuerpo monoclonal 6D12 utilizado para cuantificar el producto de glicación avanzada CML en proteínas de mitocondria de hígado de rata30,31 detecta tanto CML como CEL con diferentes afinidades (el último con mayor afinidad que el primero32) y no puede ser utilizado con la intención de cuantificar el contenido proteico en productos AGE específicos. Cabe recordar que el recurso técnico que pasa por la utilización de proteína de leche deshidratada, u otras proteínas de origen animal, para la supresión de las uniones no específicas, genera probablemente interferencias dado que constituyen por sí mismas una rica fuente de CML y otros compuestos AGE33. No obstante, cabe indicar que la inmunohistoquímica de tejidos con 6D12 y otros anticuerpos anti-AGE como el 1F6 y el 2A2 todos ellos muestran inmunoreactividad compatible con reactividad mitocondrial en tejidos humanos34.
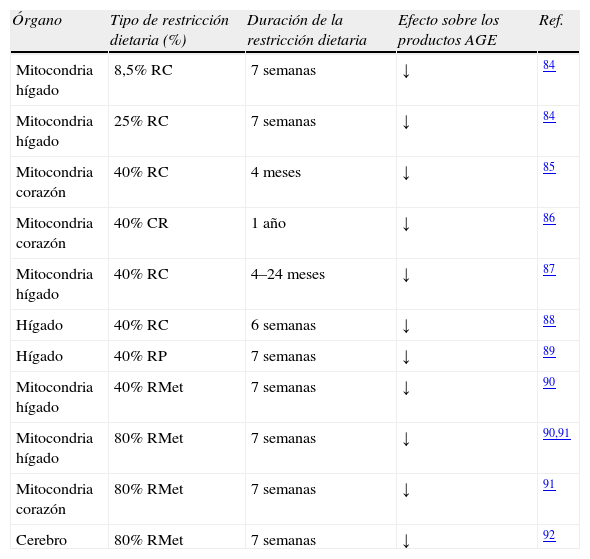
Las estimaciones de las concentraciones de AGE en proteínas mitocondriales con métodos analíticos más precisos provienen de los estudios utilizando análisis de dilución isotópica/GC-MS (tablas 1–3). En dichos trabajos se detectaron y cuantificaron 1–2mmol de CML/mol de lisina en proteínas mitocondriales de corazón de rata; por el contrario, el contenido de CML determinado por inmunoensayo con el anticuerpo 6D12 sobreestimaba aproximadamente 10 veces dicho contenido respecto al estudio mencionado anteriormente. El contenido del aducto CEL en proteínas mitocondriales era aproximadamente de 0,5mmol/mol de lisina, y el de CMC era del orden de 0,1–0,2mmol/mol de lisina. El contenido en proteínas mitocondriales de estos compuestos, CML, CEL y CMC, disminuían con la restricción calórica (tabla 5).
Efecto de la restricción calórica, proteica y de metionina en los productos de glicación derivados de metilglioxal y glioxal (CML, CEL y CMC) de diferentes tejidos de rata
| Órgano | Tipo de restricción dietaria (%) | Duración de la restricción dietaria | Efecto sobre los productos AGE | Ref. |
| Mitocondria hígado | 8,5% RC | 7 semanas | ↓ | 84 |
| Mitocondria hígado | 25% RC | 7 semanas | ↓ | 84 |
| Mitocondria corazón | 40% RC | 4 meses | ↓ | 85 |
| Mitocondria corazón | 40% CR | 1 año | ↓ | 86 |
| Mitocondria hígado | 40% RC | 4–24 meses | ↓ | 87 |
| Hígado | 40% RC | 6 semanas | ↓ | 88 |
| Hígado | 40% RP | 7 semanas | ↓ | 89 |
| Mitocondria hígado | 40% RMet | 7 semanas | ↓ | 90 |
| Mitocondria hígado | 80% RMet | 7 semanas | ↓ | 90,91 |
| Mitocondria corazón | 80% RMet | 7 semanas | ↓ | 91 |
| Cerebro | 80% RMet | 7 semanas | ↓ | 92 |
RC: restricción calórica; RMet: restricción de metionina; RP: restricción proteica.
Respecto a otros AGE, como los derivados de arginina, la cuantificación del contenido de productos AGE en proteínas mitocondriales mediante técnicas de inmunotransferencia se han llevado a cabo con uno de los mejores anticuerpos monoclonales caracterizados contra productos AGE: el anticuerpo monoclonal 1H7G5 que reconoce MG-H1, la hidroimidazolona derivada de MG35. La detección y cuantificación con este anticuerpo demostró la presencia de dicho AGE en proteínas mitocondriales de C. elegans y una disminución de la concentración de este compuesto en C. elegans transgénicos sobreexpresores de Glo11. Utilizando la misma técnica, también se demostró la presencia de MG-H1 en proteínas de mitocondrias aisladas del cortex renal de rata, estando aumentado el mismo por la diabetes36.
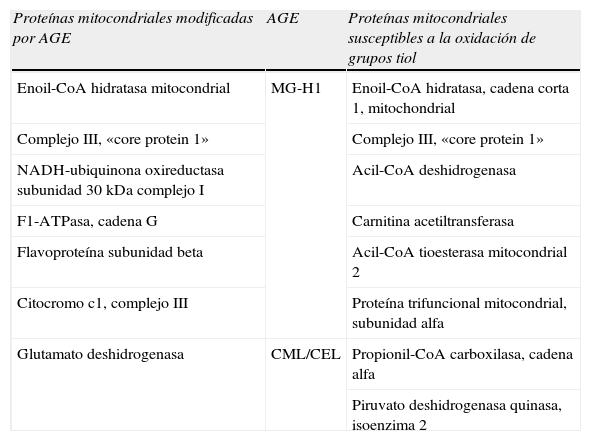
La identidad de proteínas específicas modificadas por productos AGE han sido tan solo establecidas por inmunoreactividad con anticuerpos anti-AGE, no habiendo sido corroboradas dichas modificaciones mediante análisis de espectrometría de masas. Así, se ha sugerido que la glutamato deshidrogenasa es una diana de la modificación por productos AGE mediante inmunodetección con el anticuerpo 6D1237. Otras proteínas mitocondriales identificadas por esta técnica y susceptibles de ser modificadas por MG36 se ofrecen en la tabla 6.
Proteínas mitocondriales susceptibles a la modificación por productos AGE y oxidación de grupos tiol
| Proteínas mitocondriales modificadas por AGE | AGE | Proteínas mitocondriales susceptibles a la oxidación de grupos tiol |
| Enoil-CoA hidratasa mitocondrial | MG-H1 | Enoil-CoA hidratasa, cadena corta 1, mitochondrial |
| Complejo III, «core protein 1» | Complejo III, «core protein 1» | |
| NADH-ubiquinona oxireductasa subunidad 30kDa complejo I | Acil-CoA deshidrogenasa | |
| F1-ATPasa, cadena G | Carnitina acetiltransferasa | |
| Flavoproteína subunidad beta | Acil-CoA tioesterasa mitocondrial 2 | |
| Citocromo c1, complejo III | Proteína trifuncional mitocondrial, subunidad alfa | |
| Glutamato deshidrogenasa | CML/CEL | Propionil-CoA carboxilasa, cadena alfa |
| Piruvato deshidrogenasa quinasa, isoenzima 2 |
AGE: productos de glicación avanzada; CEL: Nε-carboxietil-lisina; CML: Nε-carboximetil-lisina.
Para demostrar que la modificación por AGE del proteoma mitocondrial puede conducir a alteraciones en la función mitocondrial cabe recordar que la incubación de mitocondrias de riñón de rata con MG (10–200μM) durante tan solo 5min a 24°C produce una disminución concentración-dependiente de la respiración en estado 3, un aumento y posterior disminución de la respiración en estado 4 y una disminución del coeficiente de control respiratorio38. Estos efectos observados en un período de incubación tan corto probablemente son debidos a modificaciones que afectan a grupos cisteinil-tiol39. Así, grupos tiol reactivos ubicados en la superficie de la proteína se encuentran en péptidos de los complejos mitocondriales I, II y IV40, y se han identificado proteínas con grupos tiol potencialmente susceptibles a la modificación (Tabla 6)41. En condiciones fisiológicas, la concentración intracelular de MG es de aproximadamente 2–4μM.42 Se estima que aproximadamente un 25% del MG está ligado reversiblemente a GSH y un 50% a tioles proteicos (asumiendo una concentración intracelular de GSH y tioles proteicos de 3 y 6mM, respectivamente43; y una constante de disociación tiol/MG de 3mM3). No está claro si a concentraciones fisiológicas de MG existe un efecto agudo sobre la respiración mitocondrial, y de existir se trataría de un efecto modesto de disminución de la respiración en estado 3. Considerando los niveles basales de los diferentes productos AGE detectados y cuantificados a nivel tisular, y especialmente a nivel mitocondrial, y las consecuencias estructurales y funcionales que la modificación de proteínas por productos AGE conlleva, es más que probable que parte de los cambios fisiológicos asociados a la edad, así como en diferentes situaciones patológicas, sean debidos a la modificación irreversible por productos AGE de grupos tiol mitocondriales.
Modificación por dicarbonilos del proteoma mitocondrialExisten estimaciones que indican que el proteoma mitocondrial está constituido por unas, aproximadamente, 1.500 proteínas. De entre ellas, solo 13 proteínas implicadas en la cadena respiratoria-fosforilación oxidativa son codificadas por el genoma mitocondrial; el resto son proteínas nucleares, sintetizadas en el citosol e importadas a la mitocondria44. A día de hoy, 7 son las proteínas que se han sugerido como potenciales dianas susceptibles de formación de productos AGE (tabla 6). Se desconocen, sin embargo, las consecuencias de un incremento del grado de modificación por MG y/o glioxal de dichas proteínas mitocondriales. Enoil-CoA hidratasa está implicada en la β-oxidación de ácidos grasos. La «core protein I» del complejo III es susceptible a la S-carboximetilación y su modificación inhibe la actividad de la cadena de transporte electrónico45. Otras dianas de la modificación por compuestos dicarbonilo son diferentes subunidades del complejo I y la F1-ATPasa. La flavoproteína subunidad-β también estaba sujeta a modificación por glicación. Se sabe que la modificación de un único residuo de arginina impide la transferencia electrónica46. Por último, citocromo c1 forma parte del complejo citocromo bc1 del complejo III, una proteína citocromo tipo-c de 30kDa que se une a la membrana y que funciona como una proteína donante de electrones a la citocromo c47. Los resultados de los diferentes experimentos de incubación de glioxal y MG con mitocondrias aisladas y la aparente facilidad de difusión de dichos dicarbonilos apoyan el mecanismo postulado: la modificación del proteoma mitocondrial por dicarbonilos derivados del metabolismo contribuiría al deterioro de la función mitocondrial durante el proceso de envejecimiento y patologías asociadas.
ConclusiónLa modificación de proteínas por compuestos dicarbonilo y la detoxificación mediada por el sistema glioxalasa son factores que modulan el estado basal de estrés oxidativo celular y la actividad mitocondrial. Dichos efectos se traducen en la capacidad de modificar la longevidad de una especie. Asímismo, la capacidad potencial de generar una disfunción mitocondrial se puede traducir, en determinadas circunstancias, en unas condiciones de estrés oxidativo que puede subyacer en la aparición y progresión de diferentes patologías asociadas a la edad. La caracterización de las dianas proteicas de modificación por compuestos dicarbonilo, en especial referidas al proteoma mitocondrial, y sus implicaciones funcionales constituye uno de los retos actuales prioritarios.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Las investigaciones llevadas a cabo por los autores de este trabajo han sido subvencionadas por ayudas de I+D del Ministerio de Ciencia e Innovación (BFU2009-11879/BFI), el Ministerio de Sanidad y Consumo (RD06/0013/0012) y la Generalitat de Catalunya (2009SGR735) a RP.
Este trabajo ha recibido el Premio Pañella Casas otorgado por la SEGG.
