





La elevada prevalencia de la enfermedad de Alzheimer (EA), junto con la posibilidad de nuevas pautas en el diagnóstico mediante el empleo de los biomarcadores está cambiando el enfoque de los ancianos con demencia o en riesgo de padecerla. En este sentido parece importante revisar los aspectos genéticos de los ancianos con EA familiar, así como los ancianos con riesgo de padecer EA. La amplia difusión de los estudios genéticos asociados a esta afección también puede ser de gran ayuda. Además de los genes del amiloide, las presenilinas y la apolipoproteína E, implicados en la patogenia de la EA, debemos añadir otros genes recientemente relacionados con la enfermedad entre los que destacan el gen de la clusterina y los de fosfatidil-inositol de unión a clatrina y el del receptor de la proteína del complemento C3b.
The high prevalence of Alzheimer's disease, along with the possibility of new approaches in diagnosis through the use of biomarkers of cerebrospinal fluid is shifting the focus to the elderly with dementia or at risk. In this sense it seems important to review the genetic aspects of the elderly with familial Alzheimer's disease as well as those at risk. The wide distribution of genetic studies associated with this condition may also be helpful. To the classical findings of the genes for amyloid, the presenilins and apolipoprotein E, we must add other genes recently implicated in the pathogenesis of the disease, among which are found the clusterin gene, encoding the phosphatidyl-inositol-binding clathrin assembly protein gene, and the receptor for the complement C3b protein.
La elevada prevalencia actual de la enfermedad de Alzheimer (EA) plantea a la comunidad científica el reto de encontrar una serie de herramientas que permitan un diagnóstico lo más precoz posible de la enfermedad, con el fin de retrasar la aparición de la sintomatología cognitiva, funcional y conductual, y evitar el impacto negativo que esta afección origina en el ambiente familiar y social del paciente1. Una de las cuestiones abiertas en la demencia y la EA es un diagnóstico clínico complejo, variable y con escasa concordancia (entre el 3 y el 29%) cuando se emplean los criterios estandarizados de diagnóstico (Camdex, DSM-IV y CIE-10)2; por ello es necesaria la búsqueda de otros parámetros diagnósticos objetivos, previos a la confirmación neuropatológica para el diagnóstico definitivo.
Los avances recientes en la comprensión de la enfermedad han llevado a la comunidad científica a proponer una nueva terminología para la EA3. Para ello se ha de tener en cuenta además de la sintomatología y la evolución cognitivo-conductual, la posibilidad de complementar el estudio de los pacientes con los marcadores genéticos, de neuroimagen y de líquido cefalorraquídeo (LCR).
La aparición de esta nueva terminología3 induce a comenzar a abandonar la idea de la EA como una entidad clínico-patológica, en la que el diagnóstico no se podía asegurar desde la visión clínica, y se necesitaba la confirmación neuropatológica. Antes de esta confirmación histológica, la enfermedad ya había avanzado lo suficiente como para alcanzar el umbral clínico de demencia, y aún así, el diagnóstico solamente alcanzaba la categoría de EA probable mientras el anciano seguía vivo. El concepto que toma fuerza actualmente es la idea de una EA como entidad clínico-biológica. Con este nuevo planteamiento los biomarcadores ayudan al diagnóstico de la enfermedad en el paciente vivo, sin el concurso de los hallazgos cerebrales de la necropsia, y por supuesto antes de que la sintomatología alcance el umbral clínico de la enfermedad. En esta nueva terminología destaca la EA autosómica, o EA familiar (EAF), de carácter hereditario. Sin embargo, en la práctica clínica habitual es mucho más numeroso e importante el grupo de personas asintomáticas con aumento del riesgo de padecer EA en el futuro más o menos lejano. Nuestro análisis se va a centrar en estas 2 entidades y sobre todo en la última, por ser más prevalente. El que en la actualidad se disponga de la posibilidad de utilizar biomarcadores para el diagnóstico precoz de la EA de forma generalizada, puede tener consecuencias clínicas inimaginables en el diagnóstico y seguimiento de un proceso neurodegenerativo típico del anciano como es la EA4, ya que al poder ser detectada en estadios preclínicos iniciales, tal vez pueda modificarse mediante diversas acciones preventivas y/o terapéuticas.
La EAF la forman aquellas personas que pertenecen a las escasas familias con alteraciones genéticas, que indefectiblemente llevan a la EA por mutaciones autosómicas dominantes (también se denominada EA monogénica). Por otro lado, las personas con aumento del riesgo de padecer EA son aquellas que presentan evidencia de amiloidosis in vivo. Este fenómeno se puede detectar mediante neuroimagen o por alteración de los marcadores de LCR (Aβ42, tau y P-tau) o del plasma. También pueden presentar cambios o mutaciones en genes de riesgo, diferentes a los genes de la EAF. No obstante, en la población muy anciana, mayor de 80 años, es posible que el empleo de estos marcadores añadan pocos datos a los ya obtenidos con neuroimagen estándar y el examen neuropsicológico5.
Por otro lado, en la neurociencia actual, la investigación genética se ha desarrollado a través de la importancia que representa el relacionar los complejos fenómenos de las habilidades cognitivas, la personalidad y la psicopatología con los genes. Desde esta perspectiva, el clásico postulado reduccionista (OGOD: «one gen, one disorder»)6, solo aparece en las personas con EAF, procedentes de familias con mutaciones autosómicas dominantes, y que desarrollarán la enfermedad indefectiblemente.
La situación real, sin embargo, es bien distinta. Los trastornos de la mente y la conducta que no responden al patrón autosómico dominante son la mayoría. Estas enfermedades las podemos encuadrar dentro de las afecciones genéticamente complejas7. Bien por ser poligénicas, por presentar interacciones entre genes, o por presentar otros tipos de influencias epigenéticas que determinarán si la enfermedad se padecerá o no8. Por esta razón, la genética actual se está orientando hacia la detección de rasgos o afecciones en los que los genes tienen un papel menos drástico. De hecho, en la actualidad se buscan genes que en ningún caso son de forma aislada suficientes y necesarios para desarrollar un rasgo determinado. Esta búsqueda de genes en los que la variación alélica de un locus se puede asociar con la variación de un carácter cuantitativo, o con un aumento de riesgo de padecer una enfermedad, es la tendencia imperante en la investigación genética de la EA.
Estadios preclínicos de la enfermedad de Alzheimer y genesEnfermedad de Alzheimer monogénica autosómica dominanteLas personas que cumplen este criterio desarrollarán una EA por poseer una mutación dentro de su mapa genético, que les hará padecer la enfermedad. Estas personas son comúnmente denominadas como portadoras de EAF. Desde finales del 2010 se denomina EA monogénica o EA presintomática3. Este grupo de pacientes es muy reducido ya que su prevalencia dentro de todos los pacientes con EA supone menos del 1%9. Se han identificado hasta el momento 3 genes responsables: el gen de la proteína precursora del amiloide y los genes de las presenilinas 1 y 2 (fig. 1). La edad de inicio puede oscilar entre los 30 y los 75 años con una herencia autosómica dominante y una penetrancia completa salvo algunas excepciones10.
La proteína precursora del amiloide y el péptido amiloideo
Uno de los hallazgos típicos de la EA es el depósito del péptido amiloideo (Aβ42) a nivel extracelular11, procedente de la degradación de una proteína de membrana: la proteína precursora del amiloide (APP). A pesar de los avances en la comprensión del procesamiento de la APP como una de las bases patogénicas de la EA, las funciones fisiológicas de esta proteína todavía no están bien definidas. Estas pueden estar asociadas a cambios celulares y durante el desarrollo, sugiriendo que esta proteína es un receptor de superficie celular perteneciente al grupo de proteínas integrales de membrana tipo I con similitudes funcionales con la familia de los receptores Notch12,13.
Por otro lado, y respecto al Aβ42, este se acumula tanto en el retículo endoplásmico neuronal, como a nivel extracelular, considerándose este último fenómeno como el acontecimiento primario en la patogénesis de la EA. La acumulación del Aβ42 provoca la pérdida sináptica y la muerte selectiva de las neuronas. La hipótesis patogénica parece que resulta de la agregación de los monómeros, que normalmente no son tóxicos, en oligómeros tóxicos. Se ha confirmado que la agregación de la Aβ42 (dímeros, trímeros u oligómeros) altera la actividad sináptica dañando las espinas dendríticas, afectando la neuroplasticidad de los elementos sinápticos e inhibiendo la potenciación a largo plazo a nivel hipocampal in vivo14.
Mutaciones en el gen de la proteína precursora del amiloideEl descubrimiento del gen de la APP, situado en el brazo corto del cromosoma 21 (21p21), fue seguido por la identificación de mutaciones sin sentido asociadas con la EAF de inicio precoz15. La primera mutación sin sentido del gen APP, identificada en 199016, produce hemorragia cerebral hereditaria con amiloidosis de tipo holandés. Desde entonces se han estudiado 77 familias con 23 mutaciones sin sentido en el gen APP17. Las mutaciones se localizan en los exones (16 y 17) que codifican Aβ42 o en su proximidad, e influyen en su procesamiento, puesto que afectan a sus propiedades de producción y originan el aumento de la agregación de Aβ42. Estas mutaciones suponen menos del 0,1% de los pacientes con EA18.
Aunque se emplea el concepto de enfermedad autosómica dominante, no todos los casos son así y al menos hasta el momento, se han encontrado 2 excepciones. Son mutaciones con herencia autosómica recesiva que solo causan la EA en situación de homocigosis (deleción del trinucleótido E693Δ, y mutación A673V). Estos casos, excepcionales, explicarían la aparición de la EA a edad muy temprana y sin antecedentes familiares. También, los procesos de duplicación de la región del gen de la APP o del propio gen APP, al igual que ocurre en las personas con síndrome de Down, pueden ampliar los efectos de las mutaciones comentadas previamente. Este fenómeno de duplicación del gen origina EA temprana con angiopatía amiloidea cerebral extensa, y puede suponer hasta una quinta parte de todas las familias con EA monogénico19, aunque estas duplicaciones no siempre se han encontrado en otros grupos con EAF de inicio precoz20.
Las presenilinasLa comprensión de la función normal de las presenilinas (PSEN) y la naturaleza de sus mutaciones es fundamental para descubrir el papel de las mismas en la EA. Los genes de las PSEN codifican unas proteínas que están muy conservadas en la escala filogenética, y con una elevada homología con algunos genes de la vía Notch, siendo necesarias para la señalización del procesamiento proteolítico de proteínas transmembrana de la familia Notch. Las investigaciones realizadas definen a las PSEN como el centro catalítico de un complejo multiproteico (complejo gamma-secretasa/presenilinas) cuya disfunción origina un procesamiento anormal de los sustratos dentro de la membrana celular, que en el caso de la APP contribuye a la generación de pequeños péptidos amiloides tóxicos que desencadenan la fisiopatología de la EA21.
Gen de la presenilina 1El gen de presenilina 1 (PSEN1)22 está localizado en el brazo largo del cromosoma 14 (14q23.3), y se han identificado 393 familias con un total de 178 mutaciones diferentes. La mayoría de las mutaciones consisten en la sustitución de un único nucleótido, aunque también se han descrito pequeñas delecciones e inserciones. Estas mutaciones alteran el procesamiento de la APP, teniendo como resultado un aumento de la proporción Aβ42/Aβ40, bien por un aumento de Aβ42 o bien por un descenso de Aβ40. En gran parte, las características clínicas y neuropatológicas de la EA relacionada con PSEN1 son mucho más severas que las de la EA esporádica. La edad de inicio de la enfermedad puede ser muy temprana en algunos árboles genealógicos (hacia los 30 años). Habitualmente tiene una progresión más rápida de lo que normalmente se ve en la EA de inicio tardío, y como hallazgos neuropatológicos, una gran cantidad de depósitos de amiloide y de ovillos neurofibrilares23.
Gen de la presenilina 2El gen de la presenilina 2 (PSEN2)24 está localizado en el brazo largo del cromosoma 1 (1q31-42). Hasta el momento se han identificado sólo 23 familias con 14 mutaciones en su secuencia. Cuantitativamente es el menos importante de los tres genes implicados en la EA monogénica, y sus efectos fisiopatogénicos son similares a los descritos para el gen de la PSEN1.
El gen de la apolipoproteína E y el riesgo de padecer enfermedad de AlzheimerEn este momento podemos encontrar una serie de individuos, que sin presentar clínica de EA actual es posible que la desarrollen en años posteriores o en las edades avanzadas de la vida. Estas personas pueden presentar una serie de marcadores de amiloidosis cerebral positivos, o bien ser portadores de algún otro factor de riesgo de EA. No obstante, estos hallazgos pueden estar presentes hasta en el 30% de ancianos normales. En estos grupos poblacionales se ha comprobado mediante estudios de seguimiento longitudinales, que un porcentaje importante de los mismos acabarán desarrollando la EA25,26. No obstante, el riesgo está todavía por determinar y la susceptibilidad individual depende de otras muchas variables como: la edad del individuo, la presencia de comorbilidad, la presencia o ausencia de otros factores de riesgo o de protección (hipertensión, tabaquismo, ejercicio, dieta, entrenamiento cerebral, nivel cultural, reserva cognitiva, etc.). Alguno de ellos como la hipertensión, es posible que no tengan un papel tan claro como factor de riesgo para la EA, tal y como se ha comprobado en revisiones recientes27. A la presencia de estos factores de riesgo debemos añadir, por supuesto, el acervo genético del individuo28. La influencia genética en la EA de inicio tardío está resaltada por la presencia, hasta el momento, de un único factor claro de susceptibilidad o de aumento del riesgo: la presencia de uno o 2 alelos ¿4 en el gen de la apolipoproteína E (APOE ¿4)29.
La realización de estudios de asociación mediante análisis de ligamiento de familias con EA de inicio tardío ha permitido identificar varias regiones candidatas en los cromosomas 6, 9, 10, 12 y 21 que pueden albergar locus para genes que desencadenen EA en etapas finales de la vida30. Aunque ningún gen ha sido identificado como el responsable de los picos de ligamiento con un elevado nivel de certeza. Estas hipótesis de estudios de asociación han pasado, de análisis en los que solo se investigaron unos pocos polimorfismos de un único nucleótido (SNP) por gen, a estudios más amplios que emplean un enfoque basado en los desequilibrios de ligamiento (LD) que abarcan la variación genética completa de un gen o de una región génica, incluyendo las regiones reguladoras. Además de la hipótesis amiloidea, se analizan numerosos genes candidatos, relacionados con otras hipótesis de la EA o con otras formas de neurodegeneración (fig. 1): troceado y trasporte de la proteína APP, alteraciones en el metabolismo de la membrana y en las sinapsis, el metabolismo del colesterol, la alteración del sistema inmune, y la alteración en la regulación del calcio. No obstante, es posible que los genes relacionados con esta última hipótesis (regulación del calcio) modulen, más que la propia EA, la edad de inicio de la misma adelantándola en 2-3 años31. Como resultado de estas investigaciones se han implicado en la EA diversos genes. Sin embargo, ninguno de ellos alcanza una magnitud de efecto similar al de la APOE ¿4, en relación a su grado de asociación con enfermos o su no asociación con sanos (tablas 1 y 2). Del mismo modo, los análisis de asociación de los cluster de las regiones reguladoras del gen de la APOE o la presencia de SNP en las mismas están pendientes de replicar32. Las razones para la falta de reproducibilidad pueden incluir: insuficiente poder del estudio para detectar las variantes con contribuciones menores, heterogeneidad biológica, genética y alélica, diferencias en los diseños de los estudios y la presencia de subestructuras de la población.
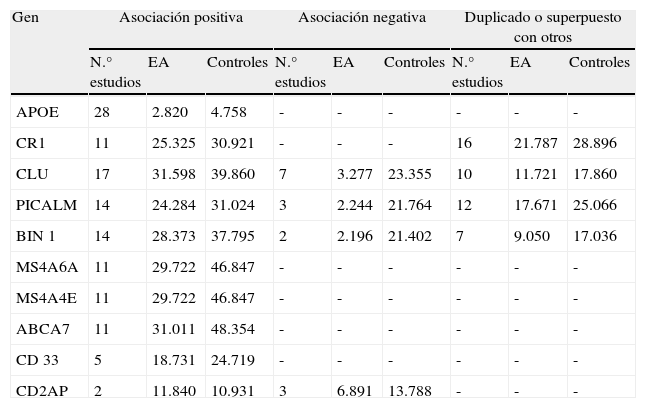
Asociación entre genes y enfermedad de Alzheimer en estudios de casos y controles
| Gen | Asociación positiva | Asociación negativa | Duplicado o superpuesto con otros | ||||||
| N.° estudios | EA | Controles | N.° estudios | EA | Controles | N.° estudios | EA | Controles | |
| APOE | 28 | 2.820 | 4.758 | - | - | - | - | - | - |
| CR1 | 11 | 25.325 | 30.921 | - | - | - | 16 | 21.787 | 28.896 |
| CLU | 17 | 31.598 | 39.860 | 7 | 3.277 | 23.355 | 10 | 11.721 | 17.860 |
| PICALM | 14 | 24.284 | 31.024 | 3 | 2.244 | 21.764 | 12 | 17.671 | 25.066 |
| BIN 1 | 14 | 28.373 | 37.795 | 2 | 2.196 | 21.402 | 7 | 9.050 | 17.036 |
| MS4A6A | 11 | 29.722 | 46.847 | - | - | - | - | - | - |
| MS4A4E | 11 | 29.722 | 46.847 | - | - | - | - | - | - |
| ABCA7 | 11 | 31.011 | 48.354 | - | - | - | - | - | - |
| CD 33 | 5 | 18.731 | 24.719 | - | - | - | - | - | - |
| CD2AP | 2 | 11.840 | 10.931 | 3 | 6.891 | 13.788 | - | - | - |
Datos de alzgene.org (2012). Resultado y número de estudios para los genes citados en población caucásica obtenidos en estudios de casos y controles (número de individuos) y su asociación con la EA (asociación positiva p<0,05).
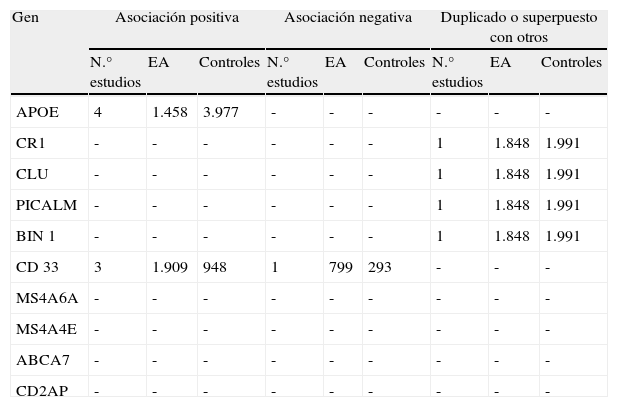
Asociación entre genes y enfermedad de Alzheimer en estudios de familias
| Gen | Asociación positiva | Asociación negativa | Duplicado o superpuesto con otros | ||||||
| N.° estudios | EA | Controles | N.° estudios | EA | Controles | N.° estudios | EA | Controles | |
| APOE | 4 | 1.458 | 3.977 | - | - | - | - | - | - |
| CR1 | - | - | - | - | - | - | 1 | 1.848 | 1.991 |
| CLU | - | - | - | - | - | - | 1 | 1.848 | 1.991 |
| PICALM | - | - | - | - | - | - | 1 | 1.848 | 1.991 |
| BIN 1 | - | - | - | - | - | - | 1 | 1.848 | 1.991 |
| CD 33 | 3 | 1.909 | 948 | 1 | 799 | 293 | - | - | - |
| MS4A6A | - | - | - | - | - | - | - | - | - |
| MS4A4E | - | - | - | - | - | - | - | - | - |
| ABCA7 | - | - | - | - | - | - | - | - | - |
| CD2AP | - | - | - | - | - | - | - | - | - |
Datos de alzgene.org (2012). Resultado y número de estudios para los genes citados en población caucásica (número de individuos) obtenidos en estudios de familias y su asociación con la EA (asociación positiva p<0,05).
Finalmente, la presencia de estudios recientes identifican hasta un 50% de enfermos en los sujetos muy ancianos homocigotos para el APOE ¿4. Esto nos orienta hacia un tipo de herencia mendeliana de carácter semidominante con un gen de penetrancia moderada, lo que implica replantearse todavía más el importante papel del APOE en la EA33.
Otros genes relacionados con el riesgo de padecer enfermedad de Alzheimer en la población ancianaLa generalización de estudios de asociación mediante el genoma completo (GWAS) permite la evaluación simultánea de millones de variantes genéticas, sin hipótesis previa, sobre posibles mecanismos biológicos. Hasta ahora se han publicado varios GWAS en relación a la EA, en los mismos aparecen diferencias en términos de diseño, aunque casi todos presentan el gen de la APOE ¿4 como el hallazgo más significativo.
Los resultados genéticos más recientes han revelado una gran cantidad de genes como posibles indicadores de riesgo de padecer la EA34, (ACAN, ATXN1, BCR, CD33, CLU, CR1, CTSS, EBF3, EPC2, FAM113B, FAM63A, GAB2, GALP, GOLM1, LMNA, LOC651924, LRAT, MS4A, MYH13, PCDH11X, PCK1, PGBD1, PICALM, TNK1, TRAK2, TRPC4AP, UBD, y los de asociación mediante el genoma completo: GWA_7p15.2, GWA_9p24.3, GWA_14q31, GWA_14q32.13, GWA_15q21.2). Algunos añaden todavía más complejidad a la EA, puesto que se trata de factores genéticos (EPC2) que pueden influir sobre los niveles de los biomarcadores y relacionar los hallazgos anormales en el LCR con el genotipo35. Son también muy recientes 3 nuevos GWAS que relacionan el gen MS4A (ampliador de 4 dominios de la subfamilia A) con la EA en población blanca europea36,37, uno de ellos realizado en nuestro país38. En las tablas 1 y 2 están recogidos los datos de los estudios de casos y controles y de las familias de población caucásica y que hasta el momento son tal vez los genes más importantes. El gen de la APOE presenta 28 estudios de asociación positiva con EA y ninguno negativo. Los genes CLU y PICALM tienen también un número importante de estudios de asociación positivos, sobre todo CLU, pero presentan también estudios negativos, así como estudios superpuestos a otros. Respecto al resto de genes y al igual que APOE, aunque con un número claramente menor de estudios, tampoco presentan estudios de asociación negativa CR1, MS4A6A, MS4A4E y ABCA7 (para una actualización de esta información, véase AlzGene en: http://www.alzgene.org/).
La utilización de los GWAS para analizar esta enfermedad, nos orienta hacia la EA de inicio tardío como uno de los desordenes genéticos complejos mejor entendidos. Estos GWAS sobre EA tardío han dado como resultado la identificación de 9 nuevos loci39: CLU (clusterina, también conocido como apolipoproteína J, o APOJ), PICALM (fosfatidil-inositol de unión a clatrina de ensamblaje proteico), CR1 (receptor de la proteína del complemento C3b), BIN 1 (integrador de unión 1), ABCA7 (transportador de unión a ATP) MS4A cluster (cluster de membrana de 4 dominios de la subfamilia A) CD2AP (proteína asociada a CD2), CD33 (inmunoglobulina similar a lectina de unión a ácido siálico) y EPHA1 (receptor de efrina 1). Estos genes de forma conjunta podrían explicar hasta el 30% de la EA de inicio tardío y señalan nuevos fenómenos relacionados con la enfermedad, con la función del sistema inmune, el metabolismo del colesterol y los procesos sinápticos en la membrana (fig. 1).
Varias de las propiedades del producto del gen CLU, una apolipoproteína abundantemente expresada en el cerebro, conectan directamente con la hipótesis amiloidea. La CLU está presente en los depósitos de Aβ42, y manifiesta una expresión incrementada en varias zonas cerebrales afectadas por la EA. Actúa como una chaperona (proteínas que participan en el plegamiento y desplegamiento de carácter no covalente de otras proteínas y macromoléculas) del amiloide, bloqueando la agregación de los péptidos Aβ42. Sin embargo, en función del equilibrio entre amiloide y CLU, esta última puede favorecer o impedir la formación de fibrillas de amiloide y la citotoxicidad, aunque se desconoce si esto mismo es lo que ocurre en los pacientes con EA.
La proteína del receptor CR1 está ligada a la EA a través de la agregación fibrilar del amiloide inducida por la activación de la cascada del complemento del C3. El Aβ42 circulante es eliminado por su adhesión al CR1 de la superficie eritrocitaria mediada por C3b. Este proceso está reducido en pacientes con EA en comparación con los individuos control. La inhibición de la vía del complemento de C3, en ratones transgénicos produce un aumento en los depósitos de Aβ42 y neurodegeneración. Esto sugiere un papel protector del sistema del complemento en los modelos murinos de la EA.
El papel exacto de PICALM en la fisiopatología de la EA no está claro, pero podría basarse en su función en el procesamiento de la proteína APP a través de una vía de endocitosis. También es posible que participe en los mecanismos de la fusión sináptica y en los procesos de formación de la memoria durante el trasporte de vesículas asociadas a algunos tipos de proteínas de membrana.
Dos de los más recientes GWAS difieren en gran medida de los anteriores40,41. En primer lugar, porque proporcionan de forma independiente una fuerte evidencia de una asociación con la CLU, apareciendo como el primer gen consistente con riesgo de padecer EA desde la identificación de APOE ¿4. En segundo lugar por el gran tamaño muestral de los mismos, que superan la barrera necesaria para detectar las variantes con un efecto menor. Junto con CLU también aparecen relacionados de nuevo con EA de inicio tardío CR1 PICALM. Sin embargo, estos 3 genes de riesgo de EA en el anciano, conjuntamente solo explican parte de la variación genética, siendo probable que exista una sobreestimación de su verdadero efecto, puesto que los cambios en estos 3 genes y su relación con el riesgo para la EA aún son desconocidos42.
El desarrollo de metaanálisis utilizando datos en bruto procedentes de los GWAS sin duda revelarán nuevos genes, debido a su mayor poder discriminatorio en la detección de alelos con un efecto menor. Por último, otro estudio43 GWA también con población muy amplia y con casos de EA incidentes, y que se ha reproducido en población independiente, ha mostrado que 2 loci: uno rs744373 cerca de BIN1 y el segundo, rs597668, cerca de EXOC3L2/BLOC1S3/MARK4, además de los previamente descritos se asociaban con el riesgo de padecer EA. Sin embargo, un modelo predictivo elaborado basándose en estos datos no resultó útil en la predicción del desarrollo de EA. Por tanto, aunque clínicamente no son aplicables, estos genes de riesgo pueden ser útiles para avanzar en la comprensión de la enfermedad y sus mecanismos etiopatogénicos, y ha sido repetidamente contrastado su papel en la EA de la población europea44.
Consideraciones finalesDebido a los últimos avances de la biología molecular, una enfermedad neurodegenerativa como la EA puede evaluarse muchos años antes que comience a producir sintomatología cognitiva. No obstante, este «diagnóstico» en fases preclínicas puede ser controvertido. Una vez excluidos los casos familiares debidos a las mutaciones de las PSEN y de la APP, en el momento actual, en ningún caso se puede realizar un diagnóstico genético de la EA. Lo que se puede ofrecer a las personas que deciden realizarse este tipo de estudios es solamente un dato relacionado con el riesgo, si bien, ningún modelo predictivo basado en múltiples marcadores genéticos ha sido debidamente validado en grandes muestras poblacionales con resultado positivo. Es cierto que presentar una homocigosis para el alelo ¿4 de la APOE puede incrementar el riesgo de padecer una EA hasta 12 veces45. Sin embargo, y debido a lo alejados que nos encontramos de los postulados deterministas clásicos de la medicina, la presencia del alelo ¿4 no es condición suficiente ni necesaria para padecer una EA en etapas tardías de la vida. Además este riesgo (¿4-EA), en contra de lo que cabría esperar, se va atenuando de manera ostensible conforme avanza la edad, con una «odds» del 20 en los menores de 75 años, que baja hasta el 10 precisamente en la población más anciana: los mayores de 75 años46. También es posible que este manejo de la información, sobre el riesgo de padecer una EA, debiera ser diferente en el ámbito investigador y en el ámbito clínico-asistencial. En este último estaría mucho más centrado con el manejo de la información, en sí misma, que con el diagnóstico. Esta problemática de cuándo, cómo y qué tipo de información se debe ofrecer a los posibles pacientes, tiene importantes implicaciones y repercusiones47, como lo ponen de manifiesto su aparición en la prensa diaria48, y revisiones recientes en esta revista49.
Además, todavía hay cuestiones no resueltas. En primer lugar, los datos relacionados con los biomarcadores bioquímicos o de neuroimagen de la EA ya nos indican que se ha puesto en marcha un proceso fisiopatológico, frente a los marcadores genéticos de pacientes en riesgo de padecer EA, que solo nos informan sobre una hipotética posibilidad de que aparezca la EA. Por este motivo, en este momento, tal vez sean más fiables a la hora de predecir riesgo de EA los marcadores morfológicos y fisiopatológicos, que los propios trastornos genéticos. Todos estos biomarcadores funcionan bien cuando trabajamos con ellos desde el punto de vista poblacional. Sin embargo, en la actividad clínica, habitualmente no se tratan poblaciones, sino personas de forma aislada. En una persona no se tiene por que cumplir necesariamente lo que ocurre en una población, esto puede ser debido a múltiples factores relacionados con la variabilidad individual y el ambiente en el que se ha desarrollado y ha permanecido el sujeto a lo largo de su vida.
Todas estas variables dan origen a la segunda cuestión: ¿cómo van a actuar los factores que pueden modular la expresión génica? Parece que van a jugar un papel determinante en la formación del fenotipo de los individuos. Actualmente los factores epigenéticos se investigan desde la biología molecular y más concretamente en relación con procesos que acontecen en las histonas del núcleo neuronal. A este nivel los aspectos epigenéticos pueden realmente influir en la expresión génica, por metilación o desmetilación de esas histonas, y determinar directamente muchos aspectos relacionados con el aprendizaje y la memoria50,51.
Tampoco podemos olvidar la plasticidad neural. Esta se mantiene incluso en poblaciones muy ancianas, y mediante los procesos de arborización dendrítica y sinaptogénesis puede influir de una manera notable en la aparición o en la velocidad de desarrollo de una afección neurodegenerativa como la EA. Al igual que la presencia de un alelo ¿4 puede ser un factor de riesgo para que aparezca la enfermedad, la estimulación de la plasticidad neural mediante el ejercicio mental puede ser un factor protector frente al deterioro cognitivo, este efecto se puede lograr de diversas formas como: actividades recreativas comunes, el ejercicio físico y modificando los estilos de vida52–55.
Por último, ¿cuál debe ser nuestra actitud terapéutica dada la situación de la enfermedad en el momento actual? Hemos de ser conscientes de que la medicación disponible para tratar a los pacientes con EA no es una medicación curativa, aunque prescrita desde fases iniciales puede ser más eficaz. En este momento no disponemos de datos de seguimiento a largo plazo de personas con la alteración de estos marcadores de riesgo (genéticos, bioquímicos y de neuroimagen) en las etapas medias de la vida. En el hipotético caso de que se planteasen ensayos clínicos de carácter terapéutico con estos grupos de personas, los primeros resultados tardarían en aparecer 30 o más años. Por este motivo, parece que los cambios en el estilo de vida como una dieta equilibrada de tipo mediterráneo, y una mayor y mejor actividad física e intelectual, probablemente sean los mejores consejos que podemos dar a la población adulta en general, y particularmente a aquellas que mayor riesgo tienen de padecer la EA: las personas ancianas.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
