



El interés en la medida de cobalto es debido al problema existente con las prótesis metal-metal. Estas liberan cobalto a la circulación sanguínea y a los tejidos produciendo efectos perjudiciales para la salud.
El objetivo de este estudio es validar un método para la medida de cobalto en suero mediante espectroscopia de absorción atómica con atomización electrotérmica y corrección de fondo por efecto Zeeman longitudinal.
Los límites de detección y cuantificación fueron de 0,31 y 1,04μg/L, respectivamente. La masa característica encontrada fue de 17,8pg/0,0044 unidades de absorbancia. La curva de calibración es lineal entre 0 y 50μg/L. La pendiente obtenida con adiciones estándar de cobalto está incluida dentro del intervalo de confianza de la curva de calibración con patrones acuosos, por tanto, no hay efecto matriz. Se comprobó la exactitud y precisión empleando material de referencia Seronorm Trace Elements Serum. La recuperación media obtenida fue del 98,97%.
El método propuesto resultó sensible, robusto, exacto y preciso para biomonitorizar la concentración de cobalto en suero como indicador de riesgo para la salud.
The interest of measuring cobalt in serum is due to the problem with metal-on-metal bearings. They release this metal into tissues and the blood circulation producing harmful effects on health.
The aim of this study is to validate a method for cobalt determination in serum samples by electrothermal atomization atomic absorption spectrometry technique (ETAAS) with longitudinal Zeeman-effect background correction.
The detection and quantification limits were 0.31 y 1.04μg/L, respectively. The characteristic mass was 17.8pg/0.0044 absorbance units. The calibration curve is linear between 0 and 50μg/L. The slope of the standard addition curve is included within the confidence interval of the calibration curve using aqueous standards, so that, there is not matrix effect. The precision and accuracy were tested using reference material Seronorm Trace Elements Serum. The mean recovery was 98.97%
The proposed method proves to be sensitive, robust, and precise for biomonitoring the concentration of cobalt in serum samples as an indicator of health risk
El cobalto es un elemento traza esencial para el organismo humano, siendo un componente esencial para la vitamina B12. Se encuentra almacenado, sobre todo, en los hematíes y, en menor cantidad, en hígado y riñones1.
En la mayoría de los pacientes sometidos a cirugía ortopédica, los implantes ortopédicos resultan biocompatibles. Sin embargo, hay un creciente reconocimiento de que, a largo plazo, estos implantes pueden estar asociados a efectos adversos sobre la salud y a ciertas respuestas de los tejidos locales en algunos de estos individuos. Estos efectos adversos están mediados por los productos de degradación de los materiales con los que se fabrican estos implantes. La introducción de prótesis metal-metal para la artoplastia de cadera ha aumentado la preocupación acerca de la respuesta biológica a los productos de degradación del metal, tanto es así que la concentración de metales en suero y orina de este tipo de pacientes resulta más elevada que en pacientes que presentan los implantes convencionales de metal-polietileno2.
En 1997, Brodner et al. demostraron que la concentración de cobalto en suero era superior en pacientes con prótesis de tipo metal-metal que en los pacientes con prótesis cerámica-polietileno3. Además, en el estudio realizado por Jacobs et al. se observó que las concentraciones de cobalto un año después de la implantación de la prótesis en pacientes de Cirugía Ortopédica fueron 6 veces superiores a las correspondientes a la precirugía2.
La liberación de cobalto a la sangre puede tener impacto sobre las funciones biológicas y celulares con posibles efectos adversos sobre el sistema inmunológico, la mutagénesis y la carcinogénesis4. Múltiples estudios han descrito síntomas sistémicos graves en pacientes con prótesis de cadera metal-metal con concentraciones elevadas de cobalto. Estos síntomas incluyen miocardiopatía e hipotiroidismo, descrito en los años 50 debido a la interferencia que ejerce el cobalto en la captación del iodo5. Por todo ello, en la actualidad existe un gran interés en la cuantificación de la liberación de cobalto a partir de prótesis como un medio para identificar las posibles reacciones adversas al metal.
La necesidad de obtener un método para la determinación de este metal se debe al reciente aumento de la demanda de solicitudes en las determinaciones de cobalto en los laboratorios clínicos de rutina por parte de los cirujanos ortopédicos6. Todo ello hace que la disposición de un método simple de absorción atómica de horno de grafito para su medida sea una ventaja para este tipo de laboratorios, especialmente en el caso de no disponer de equipos de espectrometría de masas con fuente de plasma de acoplamiento inductivo (ICP-MS).
En relación con el espécimen, la sangre total parece ser mejor debido a que requiere una menor manipulación, sin embargo, Walter et al. comprobaron que el suero es la muestra idónea para la detección de cobalto debido a que presenta un mejor reflejo de las concentraciones reales del metal que la sangre7. Además, en general, se recomienda realizar la medida del cobalto en suero debido a que existen estudios que correlacionan la concentración sérica de este metal con el desgaste de la prótesis5.
En la actualidad, no existe un punto de corte universal que defina las concentraciones de cobalto elevadas en pacientes con prótesis metal-metal. Sin embargo, se han propuesto varias concentraciones umbral, por ejemplo, la Agencia reguladora de productos sanitarios y medicinas del Reino Unido (MHRA) recomienda que la concentración de cobalto en sangre total sea inferior a 7μg/L debido a que concentraciones más elevadas indican reacciones adversas en los tejidos blandos. Otros autores establecen como punto de corte 4,97μg/L de cobalto para evitar el rechazo de la prótesis5. Smet et al. encontraron que las concentraciones de cobalto superiores a 17μg/L eran indicativas de metalosis5.
El objeto del presente trabajo es describir la metodología a seguir para la validación de un método desarrollado por el propio laboratorio para la determinación de cobalto en suero por espectroscopia de absorción atómica con atomización electrotérmica (ETAAS) con corrección de fondo por efecto Zeeman aplicable a los laboratorios clínicos.
Material y métodosInstrumentaciónPara el análisis de cobalto se utilizó un espectrómetro de absorción atómica AAnalyst 800 Perkin Elmer, equipado con horno de grafito y efecto Zeeman longitudinal, así como automuestreador AS-800 y tubos de grafito con plataforma de L¿vov integrada. También se usó una lámpara monoelemental de cátodo hueco Perkin Elmer.
ReactivosPara la construcción de la curva de calibrado se emplearon disoluciones estándares de cobalto. La calibración se realizó a partir de una disolución madre de cobalto con ácido nítrico al 2% (Perkin Elmer), cuyo valor de referencia es 1g/L. Las disoluciones patrón se prepararon por diluciones sucesivas de la disolución madre con agua bidestilada libre de metales de Versylene Fresenius.
Los reactivos químicos utilizados fueron de alta pureza, específicos para el análisis de elementos traza y con un certificado de pureza en el que figura la concentración que contienen de otros elementos.
Los reactivos utilizados para la determinación de cobalto en suero fueron los siguientes:
- –
Tritón X-100 de Sigma Aldrich.
- –
Nitrato de paladio de Perkin Elmer.
- –
Nitrato magnésico hexahidratado de Perkin Elmer.
Antes de la utilización del material de vidrio, este se limpió por inmersión en ácido nítrico al 10% (v/v) durante 24 h. Tras dicho lavado se enjuagó el material con agua bidestilada libre de metales.
Material de referenciaLas muestras de referencia en suero (2 niveles, refs. 201405 y 203105), cuyos valores de referencia son 1,2 y 3,2μg/L, respectivamente, se facilitaron por Seronorm (Billingstad, Noruega). Este material de referencia se encontraba liofilizado y se reconstituyó mediante la adición de 3mL de agua bidestilada.
Material biológicoPara evitar la contaminación se utilizaron tubos especiales para la medición de elementos traza (BD Vacuntainer® Silicón, REF 368380; 6,0mL). Se dejó que coagulara la sangre al menos 30 min a temperatura ambiente y después se centrifugaron los tubos durante 10 minutos a 1.000-1.200g8.
Modificador de matrizSe utilizó un modificador de matriz para obtener sales estables de cobalto a altas temperaturas, por lo que se eliminó el efecto matriz. Asimismo, la eliminación del efecto de matriz se comprobó mediante la realización de la curva de adiciones estándar.
La preparación del modificador de matriz se realizó de la siguiente manera:
- –
500μL de paladio (10g/L de Pd [NO3]2 en HNO3 al 15%).
- –
500μL de nitrato de magnesio (10g/L de Mg [NO3]2×6H2O).
- –
100μL de tritón X-100 (Sigma Aldrich).
- –
49mL de agua bidestilada libre de metales (Versylene Fresenius).
Las disoluciones estándar de cobalto se diluyeron con igual volumen de modificador de matriz. La dilución se realizó añadiendo a 200μL de modificador de matriz 200μL de disolución patrón, homogeneizando la muestra con la misma punta de pipeta y evitando la formación de burbujas. Las concentraciones de la curva de calibración fueron de 10, 20, 30, 40 y 50μg/L. Es importante destacar que se utilizaron estándares de estas concentraciones debido a que existen estudios que indican que las concentraciones de cobalto medias en pacientes con prótesis metal-metal con buen funcionamiento se encuentran entre 4 y 7,4μg/L. Además, en el caso de pacientes con artroplastia de cadera las concentraciones de este metal superiores a 20μg/L se han relacionado con toxicidad sistémica9,10.
La disolución blanco se preparó a partir de agua bidestilada y modificador de matriz en proporción 1:1 (v/v).
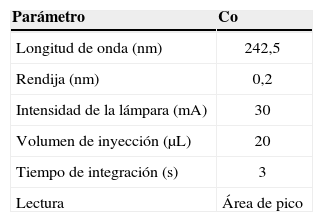
Análisis por espectroscopia de absorción atómica con atomización electrotérmicaLas condiciones instrumentales empleadas en el análisis de cobalto se esquematizan en la tabla 1.
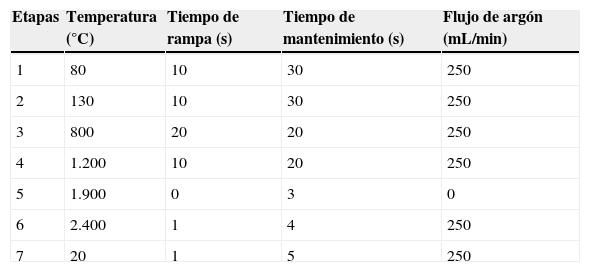
Con el fin de conseguir la mayor sensibilidad analítica posible, se utilizó un programa de temperaturas (tabla 2), que comenzó por 2 etapas de secado para evaporar el agua de la muestra. Posteriormente, y para eliminar la materia orgánica, se procedió a una etapa de pirólisis a 800°C, aumentando posteriormente a 1.200°C, con el fin de eliminar totalmente el efecto matriz. Por último, la lectura se realizó en ausencia de gas a 1.900°C, por ser la temperatura de atomización en la que se alcanzó la mayor sensibilidad analítica.
Validación del método analíticoLos parámetros característicos del método analítico se determinaron por medidas de blancos y disoluciones estándares a diferentes concentraciones. Para validar este método se determinaron los siguientes parámetros: límite de detección y cuantificación, rango de linealidad, masa característica, especificidad y exactitud, recuperación y estudio de precisión11.
Para la determinación del límite de detección, se analizaron 30 blancos acuosos y se tomó como valor del mismo el triple de la relación entre la desviación típica de las absorbancias de los blancos y la pendiente de la calibración acuosa. El límite de cuantificación se calculó como 10 veces la relación antes mencionada.
Con objeto de comprobar la linealidad del procedimiento, se realizó una curva de calibración con patrones acuosos de 5 puntos, con concentraciones desde 10 hasta 50μg/L diluidos a 1:2 con el modificador de matriz.
La masa característica se calculó utilizando la absorbancia integrada corregida por el blanco de un patrón de concentración conocida (en μg/L) sabiendo el volumen inyectado en el horno (en μL), mediante la fórmula:
concentraciónPara verificar la ausencia de interferencias provocadas por la matriz se empleó el método de adiciones estándar sobre varias mezclas de muestras reales. En este caso, se realizó una curva de calibración por el método de las adiciones estándar, adicionando a un pool de sueros cantidades de cobalto necesarias para alcanzar una concentración final de aproximadamente 10, 20, 30, 40 y 50μg/L.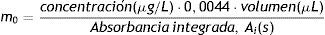
Para determinar la exactitud del método, se determinó la veracidad (expresada en términos de inexactitud), el estudio de recuperación y la precisión.
La veracidad se expresó en términos de sesgo o inexactitud (bias) y se calculó hallando la diferencia entre la media aritmética de las concentraciones halladas x¯j y el valor de referencia aceptado μ¯j12.
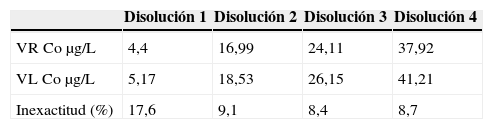
Para evaluar la inexactitud del método se analizaron 4 disoluciones de concentración 4,4; 16,99; 24,11 y 37,92μg/L de cobalto durante 10 días diferentes realizando todos los días una calibración diferente. Dichas disoluciones fueron preparadas a partir de material certificado: Seronorm Trace Elements Serum y material de referencia de Perkin Elmer, ambos citados en los apartados 2.2 y 2.3. Se aplicó la fórmula:
donde VR y VL son el valor de referencia y el valor de laboratorio, respectivamente.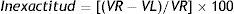
Para el estudio de recuperación se añadió a una muestra de concentración conocida, diferentes cantidades de estándar y se halló la diferencia entre la muestra analizada sin la adición de estándar y las muestras a las que se añadió estándar.
Para estimar el sesgo expresado en términos de recuperación se calculó la media teórica μ¯j que tendría que dar dependiendo de la cantidad de estándar añadida y se aplicó la fórmula siguiente:
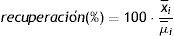
Las concentraciones empleadas para el cálculo de la recuperación fueron de 10, 20, 30, 40 y 50μg/L.
La precisión puede considerarse a 3 niveles: repetibilidad, precisión intermedia y reproducibilidad.
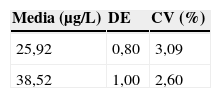
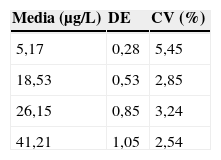
La repetibilidad se obtuvo analizando 2 pools de suero 20 veces, mientras que la precisión intermedia se halló analizando durante 10 días 4 disoluciones de concentración 5,17; 18,53; 26,15 y 41,21μg/L de cobalto realizando todos los días una calibración diferente. Dichas disoluciones fueron preparadas a partir de material certificado: Seronorm Trace Elements Serum y material de referencia de Perkin Elmer, ambos citados en los apartados 2.2 y 2.3.
ResultadosLos límites de detección y la cuantificación fueron de 0,31 y 1,04μg/L, respectivamente.
La curva de calibración con patrones acuosos de 5 puntos se observa en la figura 1. El coeficiente de variación de los factores de respuesta, relación entre la absorbancia y la concentración, fue inferior del 5% (1,01%), por lo que la calibración entre 0 y 50μg/L se consideró lineal. Además, se puede considerar que la recta de regresión pasa por el origen al estar incluido este valor dentro del intervalo de confianza del 95% del valor de la ordenada en el origen (b=–0,004-0,001). El coeficiente de correlación al cuadrado de la recta de regresión fue 0,9997.

La masa característica obtenida fue de 17,8pg y se encontró dentro del 20% del valor establecido por el fabricante (17pg/0,0044 A-s).
La pendiente de la recta de regresión de las adiciones estándar (0,002520 abs·L/μg), cuyo coeficiente de correlación al cuadrado fue 0,99355, está incluida dentro del límite de confianza del 95% de la pendiente de la regresión de la curva de calibración realizada con los patrones acuosos (0,002399-0,002560 abs·L/μg), por lo que podemos afirmar que no existe efecto matriz y que se pueden utilizar patrones acuosos para obtener la curva de calibración.
Los resultados obtenidos para evaluar la inexactitud se recogen en la tabla 3.
Se halló una recuperación media de un 98,97%.
Los resultados referentes a la repetibilidad se observan en la tabla 4, mientras que los correspondientes a la precisión intermedia se recogen en la tabla 5.
El método evaluado es lineal en las concentraciones séricas estudiadas, que comprenden desde 10μg/L hasta 50μg/L.
Debido a que valores de cobalto inferiores a 2μg/L no se consideran de relevancia clínica13, la obtención de un límite de detección de 0,31μg/L se consideró como apropiada, tratándose pues de un método sensible y adecuado para biomonitorizar la concentración de cobalto en pacientes que presentan prótesis de tipo metal-metal.
Es un procedimiento preciso y exacto utilizando material de referencia, así como muestras biológicas de suero.
Es de destacar tanto la rapidez del análisis como la ausencia de efecto matriz, lo que hace posible el análisis directo de las muestras de suero evitando cualquier procedimiento de digestión de la muestra y, por tanto, evitando también contaminaciones.
Por todo lo anterior, se considera que el método evaluado es eficaz para el análisis de cobalto en suero, lo que resulta de gran interés para los laboratorios clínicos que no dispongan de ICP-MS para biomonitorizar la concentración de cobalto en muestras biológicas como indicador de riesgo para la salud humana, especialmente en pacientes que presentan prótesis metal-metal.
Conflicto de interesesLos autores declaran no tener ningún potencial conflicto de interés relacionado con el artículo.
