




El síndrome de activación macrofágica (SAM) es una reacción patológica inflamatoria sistémica, frecuentemente fatal y comúnmente no diagnosticada, que se acompaña de una falla multiorgánica y puede desencadenarse asociada a enfermedades reumáticas, neoplásicas, infecciosas o a drogas. Más descrita en niños que en adultos, probablemente en muchas ocasiones se relaciona con alteraciones genéticas aún no descritas. Sus síntomas y signos han sido definidos. El no sospecharlo conlleva a no diagnosticarlo y como consecuencia a un incremento importante del riesgo de mortalidad en el paciente; es por esto que el diagnóstico es un reto y el tratamiento debe de ser temprano y agresivo.
ObjetivosDescribir 4 pacientes adultos con 5 episodios de SAM relacionado con diferentes enfermedades reumáticas, con el interés de familiarizar al lector con la búsqueda del síndrome y de realizar su diagnóstico.
Materiales y métodosEstudio descriptivo de pacientes adultos evaluados en la consulta y hospitalizados.
ResultadoPresentamos las características de los pacientes con SAM, el enfoque diagnóstico, las posibilidades terapéuticas y la evolución.
ConclusionesEl SAM es una enfermedad no buscada en el adulto que puede ser fatal, requiere ser identificada y tratada tempranamente para disminuir el riesgo de mortalidad. Aún requiere ser estudiada para definir defectos genéticos u otras etiologías que puedan ser responsables de este síndrome.
Macrophage activation syndrome (MAS) is a pathological systemic inflammatory reaction that is often fatal and underdiagnosed. There may be multiple organ failure that could be triggered in association with rheumatic, neoplastic or infectious diseases and/or drugs. It has been reported more in children than adults, probably as it is often associated with genetic abnormalities not described yet undescribed, genetic abnormalities. In most cases the genetic defect is not recognised in adults, or has a different aetiology. The signs and symptoms of macrophage activation syndrome have been defined. Not suspecting its presence may lead to not making the diagnosis and thus, an increase in mortality. Diagnosis is a challenge, treatment has to be started early and be aggressive to reduce the high mortality rate.
ObjectivesTo describe four adult patients with five MAS episodes related to different underlying diseases, with the aim of making it familiar to the reader, to look for the syndrome and make a diagnosis.
Materials and methodsPatients evaluated in outpatients and while in the hospital.
ResultsWe present the characteristics of MAS, with the diagnostic approach and the therapeutic possibilities and their outcomes.
ConclusionsMAS is not looked for in the adult and could be fatal. It requires identification and early treatment to reduce the risk of mortality. It still needs to be studied to define the genetic defect, or other causes that may be responsible for the development of the syndrome.
La linfocitosis familiar es una activación inmune patológica con signos y síntomas de inflamación severa, que fue reconocida en 1952 como una enfermedad de la niñez. En 1977, el síndrome de activación macrofágica (SAM) se reporta en casos aislados que presentan insuficiencia hepática y coagulopatía de consumo1, posteriormente el SAM es descrito como un problema relacionado con artritis reumatoide juvenil2. En 1993, se describe en detalle y de forma específica en 24 niños3. Hoy en día está bastante bien caracterizado constituyéndose como una entidad clínica asociada o desencadenada por drogas, infecciones, cáncer o enfermedades reumáticas o existiendo como una enfermedad aislada sin desencadenante4. El SAM ha sido descrito más frecuentemente en niños que en adultos, pero ha habido un incremento de pacientes adultos reportados5.
El SAM es un cuadro clínico similar a los síndromes hemofagocíticos, que por una deficiencia primaria o por un defecto en la apoptosis de macrófagos activados ocurre una perpetuación de la respuesta inflamatoria. Este defecto primario ha sido descrito en muchos casos, en otros su etiología aún está por definirse. El SAM ocurre cuando existe una función macrofágica mal controlada, una actividad celular citotóxica reducida y una disminución de los linfocitos T tóxicos y células asesinas naturales. Algunas citoquinas y amplificaciones de respuesta inflamatoria han sido descritas6. La apoptosis de macrófagos activados se logra por la vía de la perforina poro-granzima B serina proteasa. La reducción del número o función de células NK o una anormalidad genética del camino de la granzima-perforina llevan a un SAM.
En artritis idiopática juvenil (AIJ), lupus eritematoso y otras enfermedades reumáticas existe un incremento de la incidencia del SAM7,8. El SAM, frecuentemente se confunde con la inflamación de la sepsis o con la exacerbación de la enfermedad subyacente9. El síndrome ha sido descrito por 2 grupos diferentes, la Sociedad de Histiocitosis y por los reumatólogos pediatras. Las guías para el diagnóstico han sido publicadas por las 2 tendencias y están en proceso de definición para el lupus eritematoso sistémico (LES) y la AIJ sistémica10,11. Recientemente, los 2 grupos se reunieron y han definido la nomenclatura, así como los criterios diagnósticos12–14. El diagnóstico se basa en la presencia de criterios clínicos y criterios de laboratorio, ya establecidos por la Sociedad de Hemofagocitosis Linfohistiocítica15. Ravelli et al., elaboraron los criterios para SAM asociado a AIJ11.
Descripción: el SAM tiene un comienzo agudo y sus manifestaciones clínicas son: fiebre elevada y persistente (94%), cambios neuropsiquiátricos, esplenomegalia (59%), hepatomegalia (88%), linfadenomegalia (48%), eritema en la piel (65%), disminución del número células de 2 o 3 líneas celulares a nivel de sangre periférica, anemia (82%), trombocitopenia (88%), (>150.000 plaquetas); leucopenia (56%), enzimas hepáticas (transaminasas) elevadas (46%), fibrinógeno bajo por consumo (100%), hipertrigliceridemia (80%), tiempo de protrombina y trombina prolongados y ferritina muy elevada (100%) (en paréntesis están los porcentajes de estas características que están descritos en los criterios para SAM asociado a la AIJ por A. Ravelli11). En un artículo reciente, se reconoce la linfocitosis hemofagocítica como un desorden en incremento de diagnóstico en el adulto16.
Recientemente se ha sugerido que los niveles séricos (determinados por ELISA) del receptor específico recogedor de desperdicios (scavenger) de macrófagos el sCD163 soluble, es un marcador específico para macrófago en pacientes que tienen una activación macrofágica inadecuada, estando muy elevados y sirviendo su determinación para el seguimiento de la activación y de la actividad de la enfermedad17,18. De igual manera, la determinación de niveles del receptor soluble para el receptor de interleucina-2 (sIL-2R), se ha usado como marcador18.
En lo concerniente a la imaginología se han descrito cambios para varios sistemas y estructuras relacionadas con la respuesta inflamatoria excesiva en los órganos afectados19. La radiología de tórax demuestra un compromiso intersticial que es compatible con inflamación local o puede estar relacionado con una hemorragia intersticial. En la resonancia de cráneo se pueden ver imágenes de alta densidad o de falta de tejido en casos con compromiso del cerebro, estos hallazgos se han descrito en pacientes con síndrome de inmunodeficiencia adquirida, los hemos visto en nuestros pacientes.
A pesar de estar establecidos los criterios, aún requieren ser validados, para enfermedades diferentes a la AIJ sistémica. Se presentan situaciones que complican hacer el diagnóstico, entre otras, al comienzo de la enfermedad pudiera o no existir actividad fagocítica en la médula ósea, hígado o bazo (estos dos últimos no se consideran para las biopsias). La hemofagocitosis puede no detectarse en estas etapas tempranas y puede ser no específica en caso de una transfusión de sangre. Se puede confundir con actividad de la enfermedad subyacente o con sepsis. La elevación de triglicéridos, la determinación de células asesinas naturales y la citotoxidad de las células T, o los CD163 o los receptores para interleucina-2, no son procedimientos que se hacen de rutina en los laboratorios. La ferritina, tampoco es una prueba de rutina, ni se realiza de urgencia y pueden pasar varios días antes de obtenerse los resultados. Lo que hace que sea importante considerar el diagnóstico de SAM, como se podrá notar en los casos a continuación.
La mortalidad del SAM es alta, de 32 a 80% dependiendo de la serie revisada. La falta de conocimiento de la existencia de este síndrome, el retraso en el diagnóstico y el comienzo tardío de medidas terapéuticas pueden ser las responsables de esta alta mortalidad. Silva, presentó un número pequeño de pacientes donde coincide la alta mortalidad con el retraso en el diagnóstico, de allí la necesidad de un diagnóstico temprano20.
El virus de Epstein Barr es uno de los más descritos y hasta se lo ha responsabilizado como desencadenante. El National Institute of Health tiene una página web donde están citados los casos asociados a infecciones. Por otra parte, las enfermedades subyacentes, el carcinoma, la leucemia, la enfermedad linfoproliferativa, el LES, la dermatomiositis juvenil, la AIJ y la artritis reumatoide (AR), se asocian al SAM. Puede ocurrir con cualquier medicamento como desencadenante, la aspirina u otros antiinflamatorios no esteroideos, la segunda inyección de sales de oro, la sulfasalazina, el metotrexate21 y los anticonvulsivantes. Llama la atención que el SAM esté asociado a los biológicos anti factor de necrosis tumoral, el infliximab, etanercept o los antiinterleucina-1, el anakinra, ya que estas son las citoquinas que se disparan en los procesos de inflamación y esta última se utiliza hoy en día en muchos centros, como la terapia de elección del síndrome. Teóricamente los anti-TNF podrían ser considerados como terapia, por la misma descripción fisiopatológica del proceso, pero se ha despertado una gran discusión en ese aspecto por la paradójica respuesta descrita con estos tratamientos, algunos pacientes tuvieron la inducción del SAM con terapia anti-TNF22,23. Por otra parte, una dramática respuesta a etanercept ocurrió en un niño de 7 años de edad con AIJ24. Se describe el paciente EG, en nuestra serie que desarrolla el síndrome después de iniciado el tratamiento con anti-TNF como medicamento único25.
El tratamiento está dirigido ante todo a la eliminación de la causa que indujo el síndrome. Luego se comienza el tratamiento con pulsos de metilprednisolona a la dosis de 30mg por kg de peso o hasta un gramo al día durante 3 a 5 días, dependiendo de la gravedad del SAM y de la respuesta a las dosis iniciales. Se puede usar prednisona por vía oral a razón de uno o 2mg por kg de peso por día, en dosis única o dividida para más efecto, según la gravedad de la enfermedad. Se puede repetir el pulso en caso de respuesta insuficiente. La ciclosporina se ha usado con éxito por su efecto estabilizador de la membrana del macrófago o su función sobre los linfocitos ayudadores26,27. También se han utilizado las inmunoglobulinas y el etopósido (este último, sobre todo por la Sociedad de Hemofagocitosis). Hoy en día es muy utilizado el anakinra, que es un antagonista al receptor humano de interleucina-1 (IL-1Ra). El tocilizumab, es un anticuerpo monoclonal humanizado recombinado contra el receptor anti interleukina-6 (IL-6), se usa en el tratamiento de algunos pacientes que no toleran la interleucina-1 o no les es accesible (tabla 1).
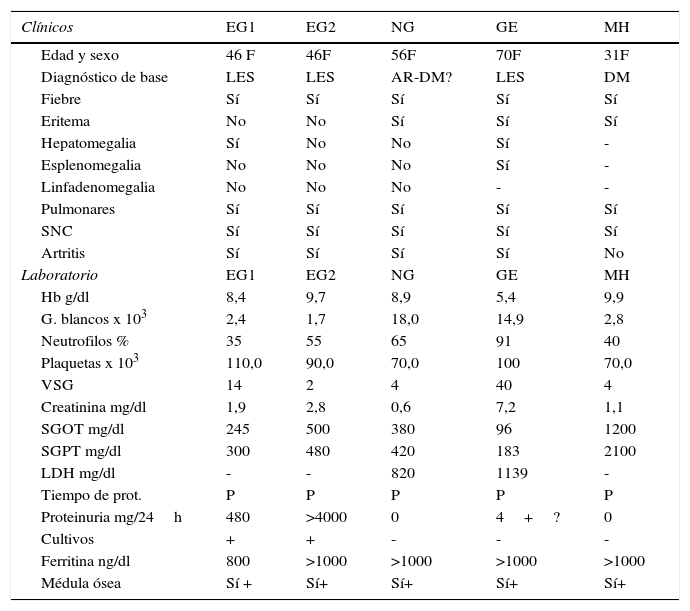
Manifestaciones y datos demográficos del SAM, en los diferentes pacientes
| Clínicos | EG1 | EG2 | NG | GE | MH |
|---|---|---|---|---|---|
| Edad y sexo | 46 F | 46F | 56F | 70F | 31F |
| Diagnóstico de base | LES | LES | AR-DM? | LES | DM |
| Fiebre | Sí | Sí | Sí | Sí | Sí |
| Eritema | No | No | Sí | Sí | Sí |
| Hepatomegalia | Sí | No | No | Sí | - |
| Esplenomegalia | No | No | No | Sí | - |
| Linfadenomegalia | No | No | No | - | - |
| Pulmonares | Sí | Sí | Sí | Sí | Sí |
| SNC | Sí | Sí | Sí | Sí | Sí |
| Artritis | Sí | Sí | Sí | Sí | No |
| Laboratorio | EG1 | EG2 | NG | GE | MH |
| Hb g/dl | 8,4 | 9,7 | 8,9 | 5,4 | 9,9 |
| G. blancos x 103 | 2,4 | 1,7 | 18,0 | 14,9 | 2,8 |
| Neutrofilos % | 35 | 55 | 65 | 91 | 40 |
| Plaquetas x 103 | 110,0 | 90,0 | 70,0 | 100 | 70,0 |
| VSG | 14 | 2 | 4 | 40 | 4 |
| Creatinina mg/dl | 1,9 | 2,8 | 0,6 | 7,2 | 1,1 |
| SGOT mg/dl | 245 | 500 | 380 | 96 | 1200 |
| SGPT mg/dl | 300 | 480 | 420 | 183 | 2100 |
| LDH mg/dl | - | - | 820 | 1139 | - |
| Tiempo de prot. | P | P | P | P | P |
| Proteinuria mg/24h | 480 | >4000 | 0 | 4+? | 0 |
| Cultivos | + | + | - | - | - |
| Ferritina ng/dl | 800 | >1000 | >1000 | >1000 | >1000 |
| Médula ósea | Sí + | Sí+ | Sí+ | Sí+ | Sí+ |
NA: no obtenido; No: ausente; Sí: presente; P: prolongado; +: positivo; -: negativo.
Edad: 46 años. Sexo: femenino. Consulta por: desorientación, hemiparesia izquierda, poliartritis y fiebre. Al examen físico está consciente, confusa y desorientada. TA: 150/110mmHg, frecuencia cardiaca 87 x minuto, peso: 40,300kg, temperatura 39,8°C, livedo reticularis severo, alopecia, 2 adenopatías cervicales no dolorosas, renitentes, no adheridas. Ruidos cardiacos apagados rítmicos sin soplos. Murmullo vesicular presente, simétrico con crepitantes bilaterales. Abdomen blando, depresible; hepatomegalia, palpable dolorosa. Bazo no palpable. Artritis en tobillo, rodilla y muñeca izquierda. Edema con fóvea en tobillo izquierdo. Fuerza muscular en miembro inferior izquierdo 3/5, miembro superior izquierdo 4/5. Reflejos osteotendinosos: 3/4 miembro inferior izquierdo; resto de los reflejos 2/4. Sin evidencia de varices.
Historia previa a la hospitalización: ocho meses previos presentaba astenia, artritis de las manos, rodillas y tobillos, pérdida de peso de 30kg, tos seca, dolor retroesternal opresivo que empeoraba con el decúbito, disnea a medianos esfuerzos, fiebre recurrente de 39 grados, trastornos de conducta y pancitopenia, diagnosticada como lupus y dengue hemorrágico. Laboratorios: Hb 8,4g/dl, GB: 2,4×103; neutrófilos 35%; plaquetas 110.000. VSG 14mm, SGOT: 245mg/dl, SGPT 300mg/dl. Creatinina 1,9mg/dl. Proteinuria 480mg/dl. Hemocultivo y urocultivo con Salmonella especies. Radiografía de tórax: infiltrado retículo nodular. Broncoscopia: hemorragia alvéolo-bronquial. Prolongación del tiempo de protrombina y del tiempo parcial de tromboplastina no medible y niveles de ferritina 800ng/ml (normal: 233ng/ml, Abbot axsym systems). El aspirado de médula ósea demuestra la presencia de macrófagos fagocitando elementos formes.
Diagnóstico de hospitalización: LES, sepsis por Salmonella, síndrome nefrótico, vasculitis cerebral, diátesis hemorrágica, hemorragia pulmonar y SAM, y se inicia tratamiento con ciclosporina, antibióticos y medidas generales con mejoría.
Dos semanas más tarde la paciente está bien y estable, no presentando síntomas, coherente, optimista, alegre, alerta, muy recuperada, con algo de debilidad y sin alteraciones al examen físico, sin artritis, con mejoría casi total de la hemiparesia izquierda y sus resultados de laboratorios muestran: hemoglobina 8,70 g/dl, GB 3.400 segmentados 65, linfocitos 33, plaquetas 300.000, creatinina 1,6mg/dl. Las transaminasas son normales. Un perfil inmunológico demuestra anticuerpo antinuclear positivo, anti-DNA positivo, valores de complemento disminuidos.
Previo a la segunda hospitalización, un mes después de la primera: la paciente suspende todos sus medicamentos y tras 7 días presenta una convulsión y fiebre. Sus valores de laboratorio son: Hb 9,7g/dl, Cr 2,8mg/dl. Glóbulos blancos 1,7×103; neutrófilos 55%, plaquetas 90×103, sedimentación 2mm, SGOT 500mg/dl, SGPT 480mg/dl; albúmina 1,8mg/dl, proteinuria >4 g/24h. Ferritina >1.000ng/ml. Se hospitaliza con una falla multiorgánica: hemorragia pulmonar, diátesis hemorrágica, insuficiencia renal severa, síndrome nefrótico e hipertensión, hemiparesis izquierda, trastornos de conducta y cognitivos. Diagnóstico segunda hospitalización: SAM, vasculitis sistémica, LES complicado con nefropatía, hipertensión arterial.
Tratamiento segunda hospitalización: esteroides, inmunoglobulinas, ciclosporina y medidas de corrección de su falla multiorgánica, se practica traqueotomía y recibe respiración mecánica con presión positiva y requiere diálisis. Tiene complicación con sepsis por Staphylococcus aureus y Escherichia coli, la paciente eventualmente no responde al tratamiento y fallece.
Caso 2 NGEdad: 56 años. Sexo: femenino. Consulta por: confusión después de convulsión.
Historia previa: seis meses antes se le diagnostica AR, recibe tratamiento con metotrexate durante un mes y por elevación de transaminasas la cambian a sulfasalazina, hidroxicloroquina y prednisona. Cuatro meses antes suspende todo y recibe tratamiento con medicamentos no conocidos, de origen natural denominados: «medicina sistémica», con el desarrollo de síntomas como dificultad para el sueño, prurito y bradipsiquia, dificultad para la deambulación, dificultad para incorporarse, enrojecimiento de color violáceo alrededor de los ojos y de la oreja con un eritema en todo el cuerpo. No tiene antecedentes de alergia o de otra enfermedad.
Examen físico de ingreso: temperatura 38,5°C. Peso: 92kg. TA: 110/60. Eritema periorbitario heliotropo, eritema descamativo en el pabellón de la oreja y un eritema en dorso de la mano y en la región anterior y posterior del tórax, tórax con buena expansión con crepitantes en base izquierda. Sin cardiomegalia, ruidos cardiacos regulares y rítmicos con taquicardia. Abdomen blando, depresible sin visceromegalia ni signos de focalización neurológica, examen articular con sinovitis de interfalángicas proximales y metacarpofalángicas. Laboratorios: hemoglobina 8,9 g/dl, con glóbulos blancos 18×103, 65% neutrófilos, plaquetas 70×103, SGOT 380mg/dl y SGPT 420mg/dl, con una prolongación del PTT, velocidad de sedimentación globular 4mm, triglicéridos elevados, 560mg/dl, LDH 820mg/dl creatinina 0,7mg/dl. Creatina-fosfocinasa 1.080mg/dl. Ferritina >1.000ng/ml. En médula ósea presencia de macrófagos fagocitando elementos sanguíneos, como se demuestra en la figura 1. Diagnóstico: AR, con hallazgos de dermatomiositis y SAM. Tratamiento y evolución: metilprednisolona un gramo durante 4 días. Se agrega ciclosporina 4mg/kg por vía intravenosa y luego dosis de mantenimiento de ambos medicamentos, con mejoría completa del cuadro clínico. Se mantuvo la ciclosporina ya que siempre que se trató de descontinuar el medicamento, la paciente presentaba signos de reagudización de SAM. La paciente se mantuvo estable y sin crisis de enfermedad con el tratamiento a base de ciclosporina.
Caso 3 GE
Edad: 70 años. Sexo: femenino. Motivo de consulta: disnea, cambios en la piel (frágil, «prensada» con pérdida de los anexos dérmicos en el antebrazo, manos, tobillos y pies), fenómeno de Raynaud y artralgias, 7 meses previos a biopsia de pulmón que demuestra fibrosis intersticial. Sin historia de hipertensión arterial, ni enfermedad renal, ni dispepsia, ni dificultad para tragar, ni calcificaciones. Alergia a medicamentos no esteroideos. Examen físico: TA: 110/70mmHg. P 80 x min R: 20 x min. El tórax tiene una expansión reducida con crepitantes en ambas bases. No tiene cardiomegalia y los ruidos cardiacos son regulares y rítmicos sin soplos ni galope. Laboratorio: son normales con la presencia de un anti sl-70. Se hace el diagnóstico de esclerodermia y se inicia terapia con prednisona 7,5mg al día, para controlar artralgias para evitar el uso de antiinflamatorios no esteroides y tomando en consideración que los esteroides pueden precipitar crisis renales en estos pacientes, cuando se utilizan a dosis elevadas. Sin mayores cambios, ni nuevos síntomas, presenta mejoría de las artralgias y después de transcurridas 2 semanas de la biopsia de pulmón. Tratamiento: metotrexate (no se pudo administrar) y etanercept.
Evolución postratamiento con etanercept: después de recibir la primera dosis de etanercept, desarrolla prurito en los brazos y hombros por un lapso de 8 h y 50 h después es evaluada por dolor abdominal y vómitos, somnolencia, anorexia, deshidratación y debilidad. Paro respiratorio, desarrolla anuria y se admite en la unidad de terapia intensiva, por falla multisistémica. Examen físico: la tensión arterial 90/60mmHg, la frecuencia cardiaca de 120 x min, la respiratoria de 40 x min, afebril. Sin lesiones nuevas en piel, ni artritis, tiene crepitantes bilaterales en ambos campos pulmonares, con un incremento del primer ruido y un soplo sistólico II/IV sin galope, el abdomen es blando con hepatomegalia. No presenta aumento del bazo. Sin signos neurológicos anormales. Laboratorios: hemoglobina 5,49 g/dl, hematocrito: 21,9%, glóbulos rojos: 2,19×103, glóbulos blancos: 14,9×103, neutrófilos: 91%, linfocitos 5%, plaquetas: 100.000/uL, velocidad de sedimentación: 40mm, glucemia: 109mg/dl; nitrógeno: 73mg/dl; creatinina: 7,2mg/dl; calcio: 8,8mg/dl; fósforo: 9,8mg/dl; colesterol: 169mg/dl; triglicéridos: 210mg/dl. Bilirrubina: 1,78mg/dl; SGOT: 96 U/l; SGPT: 183U/l; fosfatasa alcalina: 122 U/U; LDH: 1.139mg/dl; creatinina cinasa: 294 U/l; proteínas totales: 5,9 g/dl; albúmina: 2,9 g/dl; globulinas: 3 g/dl; sodio: 132mmol/l; potasio: 5,7mmol/l; cloro 97mmol/l; análisis de orina: proteínas 4+; hemoglobina 4+. Rayos X de tórax con aumento bilateral del patrón intersticial. Los cultivos de sangre fueron negativos. El aspirado de médula ósea demuestra múltiples macrófagos fagocitando elementos celulares. Diagnóstico de ingreso: falla multisistémica, SAM, hemorragia pulmonar, acidosis mixta metabólica y respiratoria.
Tratamiento: ciclosporina y metilprednisolona.
Evolución: cultivos negativos. Ferritina: 852ng/ml. Anuria, se dializa 48 h después de su hospitalización. Diátesis hemorrágica, hemorragia pulmonar y se intuba para ventilación mecánica con presión positiva. Plaquetas 60.000 y no se modifican. Melena. Tiempo de protrombina no medible. La ferritina se incrementa a 1.800ng/ml. Se agrega al tratamiento inmunoglobulina intravenosa sin respuesta clínica. La hemorragia pulmonar se reagudiza, permanece anúrica y fallece 12 días después de su ingreso.
Caso 4 MHEdad: 31 años. Sexo: femenino. Motivo de consulta: incapacidad para incorporarse y debilidad al elevar los brazos por encima de los hombros. Confusa y algo desorientada en tiempo, así como en espacio y se comporta muy introvertida y depresiva.
Diagnóstico previo: dermatomiositis, cáncer de tiroides posquirúrgico y en terapia sustitutiva.
Examen físico: TA: 120/80mmHg, pulso: 68, peso 78kg. Temperatura: 37,90C. Pápulas de Gottron, eritema periocular y heliotropo y lesiones eritematosas con mucho dolor muscular en pared abdominal y muslos. Incapacidad de incorporarse y de elevar los brazos sobre los hombros. Sin evidencia de artritis, el examen neurológico es normal, fuerza muscular conservada en pies y manos, antebrazos y piernas.
Laboratorio: hemoglobina: 11, 8 g/dl. Glóbulos blancos, 6×103, 65% neutrófilos, plaquetas 140×103, SGOT 3.800mg/dl y SGPT 4200mg/dl, velocidad de sedimentación globular 30mm, triglicéridos 200mg/dl, LDH 820mg/dl. Creatina fosfocinasa 2.800mg/dl. Tratamiento: metilprednisolona y metotrexate. Tiene un cuadro clínico posterior al tratamiento que se interpreta como reacción parecida a síndrome viral por dengue. Laboratorios: hemoglobina 9,9 g/dl, con leucocitosis, 2,8×103GB, 40% neutrófilos, plaquetas 70×103, SGOT 1200mg/dl y SGPT 2100mg/dl, sodio 128mg/dl, velocidad de sedimentación globular 4mm, triglicéridos 430mg/dl, creatinina 1,1mg/dl. Examen de orina normal. Ferritina >2000ng/dl. Médula ósea hemofagocitosis.
Diagnóstico: SAM.
Tratamiento: el metotrexato se sustituye por ciclosporina vía oral 6mg por kg de peso (200mg 2 veces al día), sin embargo, su respuesta es lenta y comienza a tener cambios relacionados a los esteroides y a la ciclosporina. Se decide agregar Mabterra, un anticuerpo monoclonal murino/humano anti CD-20, que ha sido usado en algunos casos, como recurso salvador del cuadro clínico en 21 días, se mantuvo la ciclosporina durante varios años, sin recurrencia del síndrome.
DiscusiónSe presentan 4 casos de pacientes con enfermedades reumáticas, con 5 episodios de reacción inflamatoria sistémica y con elementos diagnósticos para SAM, de los cuales todos reciben tratamiento con metilprednisolona y ciclosporina y 2 fallecen.
El primer caso EG, con LES fue subtratada para su enfermedad, presenta el SAM sin aparente desencadenante, respondiendo adecuadamente a terapia y recayendo al suspender el tratamiento, la paciente fallece. En el segundo caso NG, pareciera existir una correlación entre el uso de la «medicina sistémica» y el comienzo o desencadenante del síndrome. El tratamiento con la ciclosporina para el control de su enfermedad se hace de forma continua y permanente, después de mejorado el cuadro agudo. El caso 3 GE, enferma después del etanercept. Se atribuye a etanercept el disparo del síndrome, por la correlación causa efecto que ocurrió y por ser el único medicamento en uso25. En el cuarto caso MH, el síndrome es desencadenando por el metotrexate, ya descrito en la literatura.
El SAM se subdiagnostica. Los médicos aún no estamos familiarizados con este diagnóstico, especialmente en adultos. El diagnóstico debe de ser considerado en esos pacientes con falla multiorgánica, con síntomas de origen inflamatorio con una enfermedad autoinmune o maligna, especialmente si hay evidencia de reactivación de la enfermedad subyacente con o sin una asociación a una enfermedad viral, bacteriana o fúngica, o al uso de alguna medicación. Es importante evaluar al paciente correctamente y solicitar los exámenes descritos, para hacer el diagnóstico y administrar la terapia adecuada.
El haber hecho el diagnóstico de SAM oportunamente permite que se administre el tratamiento adecuado y se pueda disminuir la morbimortalidad por el síndrome. Algunos pacientes en edades adultas aún pueden presentar la manifestación de un problema genético. Es muy probable que definir el defecto que dispara la reacción sistémica inflamatoria sea crucial en la decisión terapéutica del futuro y en diferenciar entre los distintos cuadros inflamatorios. Si bien es cierto que el tratamiento en general es más o menos similar, la decisión del uso de esteroides, ciclosporina, etopósido, inmunoglobulina o trasplante de médula ósea es crucial en el tratamiento de SAM.
Existen muchas cosas que debemos aprender sobre el SAM, su etiología y su tratamiento.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
