






Los síndromes paraneoplásicos neurológicos se presentan en menos del 1% de los tumores sólidos y son infrecuentes en linfomas. Se asocian a tumores con alta actividad biológica y condicionan deterioro funcional y discapacidad. La dermatomiositis se asocia a cáncer, por tanto obliga al estudio de neoplasias ocultas; su diagnóstico como síndrome paraneoplásico se establece con criterios específicos. El pronóstico funcional depende del diagnóstico oportuno, control del cáncer y de la regulación de la respuesta inmunológica. Se presenta el caso de una mujer de 65 años con dermatomiositis en el curso de un linfoma B marginal variante convencional de primario cutáneo.
Neurological paraneoplastic syndromes occur in less than 1% of solid tumours and are uncommon in lymphomas. They are related to tumours with high biological activity and cause functional impairment and disability. Dermatomyositis is associated with cancer, and requires the study of hidden neoplasms. Its diagnosis as a paraneoplastic syndrome is established with specific criteria. Functional prognosis depends on early diagnosis, cancer control, and regulation of the immune response. The case is presented of a 65 year-old woman with dermatomyositis during the course of a conventional variant of a primary cutaneous B marginal lymphoma.
Los síndromes paraneoplásicos (SPN) neurológicos se presentan en menos de 1/10.000 pacientes con cáncer y se deben al efecto remoto de la neoplasia1. En tumores sólidos la frecuencia de presentación es menor al 1%, siendo los cánceres más frecuentes pulmón, mama, ovario y colorrectal; menos en páncreas, estómago, vesícula biliar, y en malignidades hematolinfoides es aún menor2,3. La incidencia de malignidad en pacientes con potencial SPN depende del tipo de desorden, varía del 5-60% y en casi el 80% de los pacientes antecede el diagnóstico de cáncer por meses e incluso años1.
La dermatomiositis (DM) es una enfermedad inflamatoria autoinmune, caracterizada por miopatía proximal simétrica y manifestaciones cutáneas1, se asocia a riesgo hasta 6 veces mayor de cáncer4, su incidencia es de 1/100.000, siendo paraneoplásica el 15-30%5, con presentación previa (40%), concurrente (26%) o posterior (34%) a las manifestaciones del cáncer1,3,4.
El diagnóstico de DM y SPN se establece con criterios específicos y el tratamiento se enfoca principalmente en el control del cáncer3 y la modulación de la respuesta inmunológica1. Se describe el curso clínico y las estrategias diagnósticas en el caso de una paciente de 65 años con DM y pérdida funcional acelerada, en quien se confirmó linfoma B marginal variante convencional de primario cutáneo; y se presentan los resultados clínicos posteriores al diagnóstico y tratamiento.
Descripción del pacienteMujer de 65 años que ingresa el 20 de mayo de 2016 por cuadro de 4 meses de síntomas constitucionales dados por fiebre vesperal, pérdida de peso, diaforesis nocturna, 2 meses de evolución de lesiones dermatológicas generalizadas tipo placa eritematosa, pruriginosa; asociadas a mialgias y debilidad muscular progresiva con pérdida de la marcha al momento del ingreso. Con antecedente de cáncer de ovario hace 19 años, tratado entonces, el cual no cursó con clínica neurológica. Al examen físico inicial, hallazgo de eritema en heliotropo, placas eritematovioláceas en tórax y miembros superiores, respetando cuero cabelludo y miembros inferiores; en valoración inicial por rehabilitación oncológica, el 24 de mayo, se identificó disfagia motora leve, debilidad muscular en las 4 extremidades de predominio proximal con fuerza muscular de 2/5 en cintura escapular y pélvica, y fuerza muscular distal de 3/5 según escala de Daniels. Ante sospecha de miopatía se procedió a determinar enzimas musculares, encontrando creatinfosfocinasa (CPK) con valor de 10552U/L. Se realizó evaluación electrofisiológica evidenciando patrón miopático con marcada denervación muscular con electromiografía cuantitativa por patrón de interferencia (IPA) que mostró un 75% de anormalidad de las unidades motoras exploradas, la cual apoyaba la sospecha clínica de miopatía inflamatoria.
Ante la sospecha de compromiso por SPN con alto impacto funcional, el 26 de mayo se consideró inicio de inmunomodulación, con seguimiento clínico funcional y control paraclínico de enzimas musculares (CPK), encontrando una disminución sutil de CPK a 8060U/L, posterior al inicio de dexametasona, en tanto se realizaban estudios diagnósticos para neoplasia. Las imágenes y marcadores tumorales descartaron enfermedad en órgano sólido y recidiva de cáncer de ovario; los estudios hematológicos reportaron biopsia de médula ósea negativa, la biopsia e inmunohistoquímica de piel del 9 de junio de 2016 aportaron resultados compatibles con linfoma B marginal variante convencional primario cutáneo; una vez confirmado el primario oncológico se definió tratamiento oncológico específico, el cual se inició el 10 de junio de 2016 con protocolo R-CVP (rituximab, ciclofosfamida, vincristina y prednisona).
Una vez iniciado el tratamiento oncológico específico, se evidenció en el seguimiento clínico de la paciente leve ganancia de fuerza muscular de miembros superiores posterior al tercer ciclo de quimioterapia, logrando mejor participación en actividades de autocuidado, asociado a un marcado descenso de la CPK al mes de iniciado el protocolo de quimioterapia (980U/L), llegando a valor de 196U/L para el quinto ciclo de quimioterapia. El 26 de octubre de 2016 se administró el sexto ciclo de protocolo R-CVP; la revaloración de la enfermedad indica respuesta completa el 18 de noviembre de 2016.
El seguimiento clínico y paraclínico evidenció una relación inversa entre los niveles de CPK y el índice de funcionalidad global de Barthel, la mejoría en el desempeño en las actividades básicas cotidianas se asoció a aumento progresivo de la fuerza muscular logrando una calificación de 4/5 según escala de Daniels, tanto en segmentos proximales como en segmentos distales; este hallazgo permite dimensionar la severidad del daño muscular y el impacto en el funcionamiento para actividades básicas; además del papel del tratamiento inmunomodulador y quimioterapéutico en la modulación del compromiso miopático (fig. 1).
 e índice Barthel (escala de funcionalidad) a lo largo de la historia de la enfermedad.")
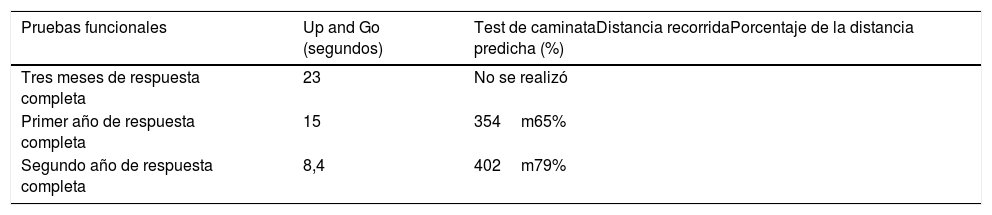
Se realizó seguimiento clínico a los 12, 24 y 27 meses de respuesta completa; además una vez la paciente recuperó la marcha, se realizaron pruebas funcionales con Up and Go y test de caminata de 6minutos, a los 3 meses de la respuesta completa, al año y a los 2 años, evidenciando ganancia en la función motora de forma progresiva, con mayor eficiencia para la marcha (tabla 1). Se mantuvo la inmunomodulación con esteroides orales y cloroquina por 18 meses; durante este seguimiento la enfermedad se encuentra en remisión completa y la paciente mantiene la independencia funcional para actividades de la vida diaria y marcha sin dispositivos.
Pruebas funcionales, test de Up and Go y test de caminata de 6minutos
| Pruebas funcionales | Up and Go (segundos) | Test de caminataDistancia recorridaPorcentaje de la distancia predicha (%) |
|---|---|---|
| Tres meses de respuesta completa | 23 | No se realizó |
| Primer año de respuesta completa | 15 | 354m65% |
| Segundo año de respuesta completa | 8,4 | 402m79% |
Dentro del amplio espectro de las enfermedades neuromusculares, que como su nombre indica abarcan desde la neurona hasta el músculo6, se deben considerar aquellas asociadas a enfermedad oncológica, agrupadas dentro de los SPN neuromusculares; estos se presentan como alteraciones funcionales del sistema nervioso, que si no se tratan de forma oportuna derivan en daño estructural permanente. Se asocian a tumores con gran actividad biológica1,7, su presentación clínica es variable y generalmente cursan con discapacidad rápidamente progresiva1,3; de ahí la importancia de su diagnóstico y tratamiento oportunos para disminuir la morbimortalidad asociada. La definición diagnóstica del SPN ha sido desarrollada por EFNS Task Force 20061 (tabla 2); clasificación basada en la presencia de anticuerpos onconeurales, el cuadro clínico y la respuesta al tratamiento oncológico e inmunomodulador con corticosteroides, azatioprina e inmunoglobulina; asimismo se recomienda el seguimiento por 5 años para pacientes sin documentación de tumor primario, en los cuales se deben realizar estudios periódicos para evaluar la presencia de malignidad, puesto que posterior al diagnóstico de SPN muchos tumores son identificados entre 4-6 meses. El riesgo de desarrollar cáncer disminuye significativamente 2 años después del diagnóstico del SPN y es muy bajo después de 4 años; sin embargo, la variabilidad clínica de estos síndromes hace difícil su reconocimiento1,2,4,5,7.
Diagnóstico de síndromes paraneoplásicos
| El síndrome paraneoplásico es definido cuando:1. Un SPN neurológico clásico (encefalomielitis, encefalitis límbica, degeneración cerebelar subaguda, neuropatía sensorial opsoclonus mioclonus, seudoobstrucción gastrointestinal crónica, síndrome miasténico de Eaton Lambert o dermatomiositis), el cáncer se desarrolla entre los 5 años del diagnóstico del desorden neurológico2. Un SPN neurológico no clásico observado y que resuelve o mejora significativamente con el tratamiento del cáncer sin inmunoterapia concomitante, que el síndrome no es susceptible a la remisión espontánea.3. Un SPN neurológico no clásico observado con anticuerpos onconeurales y cáncer desarrollado dentro de 5 años del diagnóstico del desorden neurológico4. Un SPN neurológico con anticuerpos onconeurales bien caracterizados (Hu, Yo, CV2, Ri Ma o antififisina) |
| El síndrome paraneoplásico es posible cuando:1. Un paciente con síndrome neurológico clásico no tiene anticuerpos onconeurales y no tiene cáncer pero tiene alto riesgo de un tumor subyacente2. El paciente presenta síndrome neurológico (clásico o no) con anticuerpos onconeurales parcialmente caracterizados y no cáncer3. Un paciente con síndrome neurológico no clásico sin anticuerpos onconeurales y cáncer presente dentro de 2 años siguientes al diagnóstico |
Ante sospecha de SPN, los síntomas, la edad del paciente y el perfil epidemiológico para cáncer orientan la estrategia de búsqueda; en niños se enfoca en la búsqueda de malignidad hematológica, evaluando la presencia de esplenomegalia o linfadenopatías; en los adultos, se debe explorar la presencia de tumores sólidos, requiriendo imágenes de tórax y abdomen, mamografía, ecografía testicular y colonoscopia en mayores de 50 años; se debe recurrir a la PET con fluorodeoxiglucosa (FDG-PET) cuando las imágenes son negativas, así como en la indagación de enfermedades hematolinfoides. De acuerdo con las recomendaciones de la EFNS Task Force 2006, cuando la búsqueda es negativa se debe repetir a los 3 y 6 meses durante el primer año y cada 6 meses desde el segundo año en los 4 años siguientes; posteriormente se realiza una nueva búsqueda si aparecen síntomas de alarma7.
Dentro de las teorías que buscan explicar la génesis del SPN, la teoría inmunológica es la más aceptada, afirma que el tumor expresa antígenos, denominados onconeurales, asociados con tipos específicos de cáncer, con una especificidad del 90%. Sin embargo, menos del 50% de los pacientes presenta anticuerpos onconeurales, solo el 5-10% anticuerpos atípicos no caracterizados y hasta un tercio de los pacientes no los presenta. Los pacientes con linfoma pueden presentar SPN en ausencia de anticuerpos onconeurales, de manera que esta teoría no explica la totalidad de los cuadros neuromusculares y la ausencia de estos anticuerpos no descarta malignidad1,2,7,8.
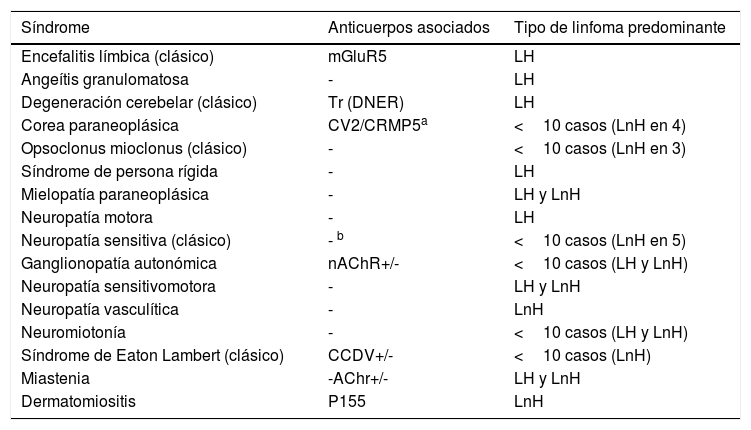
En linfoma, los SPN neurológicos se presentan de forma esporádica4. Según el consorcio europeo de SPN Euronetwork, la asociación de un SPN específico varía entre el linfoma de Hodgkin y el linfoma no hodgkiniano (tabla 3); sin embargo, de manera global la incidencia de SPN es mayor en linfoma de Hodgkin2. La asociación con DM es mayor en linfoma no hodgkiniano, mientras que la polimiositis ha sido descrita en ambos tipos de linfoma2.
Síndromes neurológicos paraneoplásicos y tipo de linfoma predominante
| Síndrome | Anticuerpos asociados | Tipo de linfoma predominante |
|---|---|---|
| Encefalitis límbica (clásico) | mGluR5 | LH |
| Angeítis granulomatosa | - | LH |
| Degeneración cerebelar (clásico) | Tr (DNER) | LH |
| Corea paraneoplásica | CV2/CRMP5a | <10 casos (LnH en 4) |
| Opsoclonus mioclonus (clásico) | - | <10 casos (LnH en 3) |
| Síndrome de persona rígida | - | LH |
| Mielopatía paraneoplásica | - | LH y LnH |
| Neuropatía motora | - | LH |
| Neuropatía sensitiva (clásico) | - b | <10 casos (LnH en 5) |
| Ganglionopatía autonómica | nAChR+/- | <10 casos (LH y LnH) |
| Neuropatía sensitivomotora | - | LH y LnH |
| Neuropatía vasculítica | - | LnH |
| Neuromiotonía | - | <10 casos (LH y LnH) |
| Síndrome de Eaton Lambert (clásico) | CCDV+/- | <10 casos (LnH) |
| Miastenia | -AChr+/- | LH y LnH |
| Dermatomiositis | P155 | LnH |
CCDV: canal de calcio dependiente de voltaje; LH: linfoma de Hodgkin; LnH: linfoma no hodgkiniano; nAChR: receptor nicotínico de acetilcolina.
No presente en todos los casos.
b Un caso con linfoma no Hodgkin y anticuerpos Ma2 (no publicado).
+/-: característico del síndrome, no predictor de cáncer.
Tomado de Graus et al.2.
Respecto al compromiso muscular en SPN relacionado con linfoma, ocurre en mayores de 50 años, con mayor frecuencia asociado a DM, hasta en un 50% de los casos la miositis antecede el diagnóstico de malignidad linfoide, a diferencia de los SPN asociados a tumores sólidos en los cuales se presentan en estadios avanzados del cáncer2.
La DM es una miopatía autoinmune, cuya incidencia varía de 0,5-0,89 por 100.000/año, con relación mujer hombre de 2:1. Aunque puede presentarse a cualquier edad se han descrito 2 picos de incidencia entre 5-15 y 45-65 años3,4,8. Como SPN, se presenta en el 7-30% de los casos9 y se diagnostica con los criterios clínicos y paraclínicos de Bohan y Peter5,8,9 presentados en la tabla 4. En la clínica se caracteriza por debilidad muscular progresiva y manifestaciones cutáneas. La elevación de enzimas como la CPK es el marcador más sensible de daño muscular1,3–5,8,9. El hallazgo de unidades motoras polifásicas, de pequeña amplitud y corta duración, descargas repetitivas, ondas agudas positivas y fibrilaciones en la exploración electromiográfica confirma el compromiso funcional intrínseco de la fibra muscular. A su vez, la biopsia muscular evidencia el daño estructural e infiltración mononuclear, aportando información clave para la discusión de diagnósticos diferenciales. Ambos estudios precisan la correcta selección del músculo a evaluar, además de su correcta interpretación. En cuanto a la presencia de anticuerpos onconeurales específicos para esta condición, aún no han sido descritos7.
Criterios de Bohan y Peter
| Criterios clínicos | Criterios paraclínicos |
|---|---|
| 1. Debilidad muscular proximal simétrica5. Signos dermatológicos (erupción en heliotropo, poiquilodermia, pápulas de Grotton y dermatitis en codos, rodillas o pies) | 2. Biopsia muscular con evidencia de miositis3. Aumento sérico de enzimas musculares4. Patrón electromiográfico característico (unidades motoras polifásicas, de pequeña amplitud y corta duración, con descargas repetitivas, ondas agudas positivas y fibrilaciones (2) |
| Categorías diagnósticas- Definitivo: 5 más 3 de cualquiera de los criterios 1 a 4- Probable: 5 más 2 de cualquiera de los criterios 1 a 4- Posible: 5 más uno de cualquiera de los criterios 1 a 4 | |
El primer caso de DM asociado a cáncer de estómago se reportó por Stertz en 19164. La tasa de incidencia de tumores en pacientes con DM es variable, de 0,6-10 por 100.000 individuos8. El estudio de Souza y Shinjo indicó que de 139 pacientes con DM el 8,6% tuvo diagnóstico de cáncer en el primer año10, mientras el estudio de Zhang et al. encontró que entre 678 casos de DM con seguimiento durante 34 años, se reportó malignidad en 115 casos (17%)11. Ante el diagnóstico de DM se debe buscar la presencia de malignidad, por la mayor morbilidad y alto riesgo de mortalidad. Son factores de riesgo para sospechar asociación de DM con cáncer: edad avanzada, género masculino, curso con disfagia, vasculitis cutánea, necrosis de piel, curso acelerado de la enfermedad4,10,11. Las neoplasias malignas más frecuentemente asociadas con DM son el cáncer de ovario y de mama en mujeres, y el cáncer de pulmón en hombres4,9; sin embargo, debe considerarse que la asociación con un tipo de tumor específico es variable; por ejemplo Zhang et al. reportaron el carcinoma nasofaríngeo como el más frecuente (51,3%), lo cual se relaciona con el perfil epidemiológico de este grupo dada la alta incidencia de esta malignidad en la población asiática11. Los tumores malignos cutáneos también se asocian con enfermedad autoinmune del tejido conectivo incluyendo DM y polimiositis4.
La DM puede ser previa (40%), concurrente (26%) o posterior (34%) al diagnóstico de cáncer1,3,4. Existe un alto riesgo en los siguientes 5 años para la presentación de cáncer de ovario, páncreas y pulmón; el riesgo de linfoma es alto solo durante el primer año después del diagnóstico; para otros tumores el riesgo es mayor dentro del primer año de seguimiento y disminuye sustancialmente después7.
El enfoque terapéutico de los SPN se orienta hacia la inmunomodulación con corticosteroides y azatioprina, y el estudio de la malignidad para el inicio del tratamiento oncológico de forma oportuna1,5,7; el uso inmunoglobulinas intravenosas y plasmaféresis ha tenido resultados clínicos favorables en pocos casos1. Aproximadamente un 30% de los pacientes queda con discapacidad8; se consideran factores de pronóstico funcional en el paciente con DM la edad, severidad de la enfermedad muscular y el compromiso sistémico asociado, la rápida respuesta a corticoides se asocia con pronóstico favorable9.
Considerando en nuestra paciente el antecedente de cáncer de ovario y la presencia de linfoma cutáneo, se buscó acerca de la frecuencia de SPN en segundos tumores primarios en la literatura, encontrando reportes de segunda malignidad tanto sincrónicos como metacrónicos. Sakai et al. reportan un caso de DM en cáncer metacrónico de la mama contralateral e indicaron que revisaron 20 casos de cáncer de mama asociados con DM, esto en población asiática12. Por su parte Voravud et al. reportaron 6 casos de cáncer de seno asociado a DM, en 2 de los cuales se confirmó carcinoma primario de ovario como segunda neoplasia, e indicaron que la DM que se desarrolla durante el curso del cáncer de seno puede indicar la aparición de una segunda neoplasia maligna primaria o cáncer de seno recurrente13. A pesar de que la malignidad dual es raramente asociada a SPN, Nasri et al. en 2016 resumen los reportes de caso de malignidad dual y SPN, sin incluir DM, o casos de SPN y linfoma14. Se encontró un caso de una paciente con 3 primarios, dados por cáncer metacrónico bilateral de mama y en el curso de DM la confirmación de coexistencia con adenocarcinoma de endometrio; indican además en este reporte que la ocurrencia de DM en el contexto de malignidad múltiple es raro15. No se encontró ningún caso de DM paraneoplásica en el contexto de linfoma o cáncer de ovario.
En el caso clínico presentado, la paciente cumplía los criterios clínicos para un diagnóstico definitivo de DM, por la elevación marcada de CPK, hallazgos electromiográficos y clínicos; no se realizó biopsia de fibra muscular dado el compromiso clínico de la paciente que limitó su realización. Se trata además de un caso de SPN clásico definido cuya evolución mostró disminución de la destrucción de la fibra muscular con el tratamiento inmunomodulador y recuperación motora y funcional progresiva con el tratamiento oncológico específico. Una vez lograda la respuesta completa de su enfermedad oncológica, continuó mejorando hasta alcanzar la independencia funcional, que se ha mantenido sin deterioro a los 2 años de seguimiento (fig. 1). Se encuentran factores pronósticos favorables derivados del diagnóstico oportuno tanto del SPN como de la malignidad hematolinfoide, que permitieron el inicio oportuno de la inmunomodulación y el tratamiento sistémico, mitigando la pérdida muscular y la recuperación funcional; sin embargo, su desempeño en el test de caminata, a pesar de la mejoría durante el seguimiento clínico, persiste por debajo de lo esperado.
ConclusiónLa sospecha de un SPN neurológico obliga a la búsqueda activa de malignidad. El presente caso describe un cuadro de DM paraneoplásica como presentación inicial en linfoma cutáneo. No se realizó biopsia muscular ni anticuerpos onconeurales, sin embargo, este es un caso de SPN clásico definido, lo que da valor a la evaluación clínica y electrofisiológica por profesionales con experiencia específica en cáncer. El grado de destrucción de la fibra muscular se asocia a la actividad biológica del cáncer y ocasiona mayor morbilidad y discapacidad, y aunque la respuesta a esteroides puede considerarse un factor de buen pronóstico, la clave de su tratamiento es el tratamiento oncológico específico.
FinanciaciónNinguna
Conflicto de interesesLas autoras no tienen conflictos de interés que afecten la información presentada en este caso clínico.
Agradecimientos a Miguel Moreno Capacho, médico especialista en Medicina física y rehabilitación; fisiatra, especialista en rehabilitación oncológica; jefe, Servicio de rehabilitación, Instituto Nacional de Cancerología E.S.E., Bogotá, D.C. Colombia por su colaboración en la revisión del presente manuscrito.
