










La hipercolesterolemia familiar (HF) es una alteración de origen genético que clínicamente se puede manifestar desde el nacimiento y que se caracteriza por niveles plasmáticos anormalmente altos de colesterol LDL (cLDL) y por una elevada tasa de morbimortalidad cardiovascular prematura. Tiene dos formas de presentación: la HF heterocigótica (HFHe) y la HF homocigótica (HFHo); esta última más severa y de aparición clínica en los primeros años de vida. Históricamente, la prevalencia para la HFHe es de un caso en 500 personas y para la HFHo de un caso por cada millón de personas; sin embargo, los datos reales probablemente son superiores porque hay evidencia de que ambas condiciones están subdiagnosticadas. La terapia recomendada, además de los cambios en el estilo de vida, son las estatinas; sin embargo, con estos fármacos es difícil lograr en muchos casos reducciones aceptables del cLDL, por lo que se requiere asociar otras modalidades terapéuticas, algunas de ellas recientemente aprobadas. Dado que en Colombia no se ha publicado ningún documento de revisión sobre HF, la Sociedad Colombiana de Cardiología y Cirugía Cardiovascular convocó a diferentes especialidades de la medicina para elaborar un documento sobre el tema, que resumiera, de manera práctica y actualizada, aspectos clínicos, genéticos, diagnósticos y de tratamiento.
Familial hypercholesterolemia (FH) is a genetic disorder that may clinically manifest since birth and is characterized by abnormally high plasma LDL cholesterol (LDLc) levels and a high early cardiovascular morbidity and mortality rate. FH has two presentation forms: heterozygous FH (HeFH) and homozygous FH (HoFH), the latter being more severe and with a clinical onset during the first few years of life. Historically, HeFH prevalence is of 1:500 and HoFH of 1:1 million; however, real data are probably higher because evidence indicated that both conditions are underdiagnosed. Recommended therapy, besides lifestyle changes, are statins; nevertheless, these drugs make it difficult in many cases to achieve reasonable cLDL reductions, therefore an association with other therapeutic models, some of which have recently been approved, is required. Since no review papers have been published in Colombia regarding FH, the Colombian Cardiology and Cardiovascular Surgery Society invited several medical specialties to draft a document on the subject that would sum up, in a practical and updated way, clinical, genetics, diagnostics and therapeutic aspects.
La hipercolesterolemia familiar (HF) es una enfermedad genética, caracterizada por niveles plasmáticos anormalmente elevados de cLDL y por una alta tasa de morbimortalidad por enfermedad cardiovascular, principalmente coronaria, en edades tempranas1–12. La HF se transmite de forma autosómica dominante, y la causa más frecuente son mutaciones en el gen que codifica los receptores para la LDL (RLDL), mientras que es menos frecuente por mutaciones en el gen de la apolipoproteína B100 (ApoB 100), en el gen de la proproteína convertasa de subtilisina/kexina 9 (PCSK9) o en el gen de la proteína adaptadora 1 del receptor de LDL (LDLRAP1)13–15.
La HF se puede manifestar de manera homocigótica (HFHo) cuando se heredan los dos alelos con mutaciones en alguno de los genes responsables, o de manera heterocigótica (HFHe) si solo uno de los alelos tiene la mutación y el otro es normal. La HFHo se caracteriza por niveles de cLDL extremadamente altos, signos clínicos por depósitos subcutáneos de colesterol (en piel, tendones y córnea), enfermedad cardiovascular, principalmente coronaria, y compromiso valvular aórtico, generalmente entre los 10 y 30 años de edad. La HFHe se caracteriza por niveles plasmáticos de cLDL habitualmente no tan altos como en la HFHo, pero en general superan los 190mg/dL, son menos frecuentes los signos clínicos, y los eventos coronarios se observan en edades entre los 30 y 50 años. En ambos casos, puede haber historia familiar de enfermedad coronaria prematura1–12.
La prevalencia de la HFHo se ha estimado en un caso por millón de habitantes, en tanto que la de la HFHe en un caso por cada 500 personas; sin embargo, se han publicado datos que sugieren que puede ser mayor16. Aunque el diagnóstico puede ser clínico en un alto porcentaje de los casos utilizando distintos criterios clínicos y de laboratorio, el diagnóstico definitivo se logra con el estudio genético17–19. En el tratamiento de la HF los cambios terapéuticos en el estilo de vida son importantes, pero necesariamente se deben incluir fármacos como las estatinas; sin embargo, debido al defecto molecular no siempre se logra una reducción eficiente de las concentraciones de cLDL con monoterapia y por ello se requiere asociar otros hipolipemiantes como ezetimibe o anticuerpos monoclonales contra la PCSK9, estos últimos recientemente aprobados por la Administración de Drogas y Alimentos (FDA, su sigla en inglés) y la Agencia Europea de Medicamentos (EMA, su sigla en inglés) para estos pacientes, pero aún no en Colombia. El tratamiento de la HFHo es todavía más difícil; además del tratamiento combinado de estatinas y ezetimibe, actualmente se dispone de fármacos como lomitapide, mipomersén, anticuerpos monoclonales anti-PCSK9 y terapia no farmacológica como la aféresis de LDL2–12.
Necesidad de un documento de revisión sobre hipercolesterolemia familiarA pesar del alto impacto social y económico de esta condición21, no hay una publicación en Colombia dirigida al diagnóstico y tratamiento de la misma. Por ello, la Sociedad Colombiana de Cardiología y Cirugía Cardiovascular convocó a especialistas en Cardiología, Medicina interna, Pediatría, Endocrinología y Genética para elaborar un documento de revisión que resumiera de manera práctica y actualizada los aspectos clínicos, genéticos, diagnósticos y de tratamiento de la HF, y así mismo permitiera aumentar el conocimiento e interés de la población médica. Como objetivo principal se busca prevenir de manera temprana la aterosclerosis prematura, pero también, aumentar el interés en la detección de las manifestaciones de la HF, promover la investigación nacional y estimular la creación de registros y bases de datos epidemiológicas y clínicas, para lo cual se busca como propósito la puesta en marcha de un Registro colombiano de HF utilizando una base estructurada internacional ya existente, que permita no sólo el conocimiento detallado de las frecuencias y distribuciones de las formas de HF, sino el intercambio de datos con otros países, en particular con la región iberoamericana (www.colesterolfamiliar.org/estudio-safeheart). Este documento será particularmente útil para médicos generales, pediatras, internistas, cardiólogos, endocrinólogos, genetistas, dermatólogos y especialistas en medicina familiar, por ser estas las especialidades de mayor contacto con los pacientes con HF.
Aspectos genéticos de la hipercolesterolemia familiarGen del RLDLAproximadamente el 90% de los casos de HF son causados por mutaciones o variantes funcionales en el gen del RLDL13–15. El gen del RLDL se localiza en el cromosoma 19 y hasta la fecha se han descrito más de 1.700 mutaciones25,26. La HF se puede manifestar de manera homocigótica (HFHo), cuando ambos alelos (uno de cada progenitor) del mismo gen, tienen la misma mutación (HFHo verdadero), o de manera heterocigótica (HFHe) si se hereda solamente un alelo con mutación de uno de los padres. Se pueden heredar mutaciones distintas en ambos alelos del mismo gen (HF heterocigótica compuesta), o menos frecuentemente pueden heredarse de ambos padres mutaciones en genes diferentes (heterocigóticos dobles)9,13. De acuerdo con el grado de actividad residual del receptor del LDL, las mutaciones en el gen RLDL se han clasificado convencionalmente como mutación de alelo nulo si la actividad es menor del 2%, o de alelo defectuoso si la actividad está entre el 2 y el 25%22. Los pacientes homocigóticos con mutaciones de alelo nulo suelen tener valores plasmáticos de cLDL extremadamente altos, y pronóstico y tratamiento desfavorables23,24. En los pacientes con HFHe, la expresión clínica dependerá del tipo de mutación y del grado de actividad del receptor dependiente del alelo sano.
Gen de la ApoB 100Este gen se localiza en el brazo corto del cromosoma 2. Representa entre el 5 al 10% de los casos con HF. Las mutaciones en este gen producen una proteína con una capacidad de unión al RLDL limitada, lo que conduce a un incremento del cLDL27.
Gen de la PCSK9Este gen se encuentra en el brazo corto del cromosoma 1 y codifica una proteína que se une al complejo RLDL-ApoB100, favoreciendo la degradación intracelular del receptor. Representa entre el 1 y el 3% de los casos de HF. Se han demostrado dos mutaciones de ganancia de la función de la PCSK9 que invariablemente aumentan los niveles plasmáticos de cLDL (28) y una forma de mutación que genera reducciones importantes en el cLDL (pérdida de la función de la PCSK9)29.
Gen de la LDLRAP1Está localizado en el brazo corto del cromosoma 1; codifica la proteína adaptadora 1 del RLDL. Las mutaciones de este gen en homocigóticos producen un cuadro similar a la HF homocigótica conocida como hipercolesterolemia autosómica recesiva y representa menos del 1% de los casos30.
Asesoría genéticaLas personas con diagnóstico definitivo de HFHe tienen a uno de sus progenitores afectado, y la probabilidad de transmitirla a su descendencia es del 50%. Si ambos progenitores padecen de HFHe, la probabilidad de que sus hijos hereden la condición heterocigótica es del 50%, mientras que la homocigótica es de un 25%. Cabe anotar que hasta 25% de los hijos pueden tener valores normales de colesterol.
Mecanismos fisiopatológicos de la hipercolesterolemia familiarTransporte de lípidosLas células necesitan colesterol y grasas para crear y mantener la membrana celular y para organizar su estructura interna, así como para la producción de las hormonas esteroideas. Los lípidos no son solubles en el plasma y necesitan transportadores, denominados lipoproteínas, que a su vez tienen proteínas externas, llamadas apoproteínas, cuya función principal es servir como ligandos con receptores específicos31.
Control de los niveles séricos e intracelulares de colesterolLa síntesis y el uso del colesterol necesitan una regulación estricta para que esté disponible en sus funciones, pero también para evitar su exceso32. Todas las células están en capacidad de hacer esta regulación. El control de niveles de colesterol depende de dos aspectos principales: de la producción intracelular de colesterol y de la captación de colesterol por la célula.
Síntesis de colesterolLa producción depende de varios pasos y en el paso entre hidroximetil glutaril coenzima A y mevalonato interviene el NADPH como agente reductor, pero depende principalmente de la reductasa de HMG CoA, en una reacción que es limitante de la velocidad de síntesis del colesterol. Esta enzima es el objetivo del tratamiento farmacológico con estatinas. Cuando la estatina actúa se reduce la actividad de la reductasa y disminuye la producción intracelular de colesterol, al tiempo que aumenta secundariamente la actividad de los receptores específicos para LDL (fig. 1).
Captación del colesterol
La célula tiene control sobre los niveles de colesterol gracias a dos mecanismos principales: la expresión de la reductasa de HMG CoA (puede aumentar o disminuir la síntesis de colesterol), y la captación de colesterol extracelular. El proceso de captación de colesterol necesita la expresión de un receptor específico, RLDL, y la actividad de una proteína adaptadora para el RLDL (LDLRAP1), que interviene en el ingreso a la célula del receptor, una vez que este ha captado el colesterol33.
El receptor RLDL, con ayuda de la proteína LDLRAP1, toma el colesterol y facilita la endocitosis. El RLDL es una proteína que se expresa en la superficie externa de la célula y que actúa como receptor para la ApoB, que está en la superficie externa de los fosfolípidos de la LDL. El RLDL también actúa como receptor para la ApoE que está en la superficie de los remanentes de quilomicrones y en las IDL. Cuando se unen el receptor y la LDL se produce una invaginación de la membrana, que luego se cierra y fusiona, formando una vesícula intracelular recubierta de clatrina, que lleva el LDL al interior de la célula a su destino intracelular34. Este proceso ocurre en todas las células con núcleo, pero principalmente en el hígado, que retira más del 70% del LDL de la circulación. Una vez que la vesícula ha ingresado al medio celular, el cambio en el pH produce un cambio conformacional en el receptor, que libera la partícula de LDL y el receptor vuelve a la superficie celular, en la que el pH neutro le devuelve su conformación original y lo dispone para recibir otra partícula de LDL.
Control de los niveles plasmáticos de colesterolLa síntesis de RLDL depende básicamente del nivel de colesterol libre intracelular. Cuando hay exceso de colesterol libre se inhibe la transcripción del gen del receptor. Hay otro mecanismo para la regulación del colesterol, mediante la destrucción del RLDL antes de su salida a la superficie celular. Una proteína sintetizada en el interior de la célula, la proproteína convertasa de subtilisina-kexina (PCSK9) sale al exterior y se une al RLDL. Cuando el complejo de PCSK-9 y RLDL+LDL ingresa a la célula se libera el LDL, y la PCSK9 induce la degradación del receptor35. Cuando hay ganancia en la función del gen PCSK9 se disminuye el número de receptores para LDL en la superficie celular, con dos consecuencias: al no haber ingreso de cLDL aumentan los niveles plasmáticos, y la falta de colesterol en el interior de la célula, interpretada como déficit por el núcleo, se traduce en aumento en la expresión de la reductasa de la HMGCoA, que lleva a aumento en la síntesis de colesterol y, por tanto, de las concentraciones plasmáticas.
Mecanismos fisiopatológicos de las dislipidemiasHay dislipidemias secundarias (diabetes mellitus, hipotiroidismo, síndrome nefrótico, medicamentos o hábitos de vida). Las dislipidemias primarias, de origen genético, producen dislipidemia de acuerdo con lo expuesto previamente36. La figura 2 muestra las causas genéticas más frecuentes de hipercolesterolemia.
Alteración en el gen RLDL
El defecto primario en la cantidad de los receptores o en la calidad de su actividad se traduce en déficit en la captura de colesterol. El déficit en la conformación del RLDL lleva a la dificultad o imposibilidad para tomar el LDL, con la consecuente elevación de los niveles lipídicos.
Alteración en el gen ApoBLa LDL se une al RLDL mediante el ligando, que es la ApoB, la cual puede estar alterada cuando el defecto está en el gen que codifica su expresión37.
Alteración en el gen PCSK9Como mecanismo interno de regulación del colesterol, la PCSK9 disminuye la recirculación de los RLDL porque estimula su degradación una vez que ha liberado el LDL en el interior de la célula28. La ganancia en función del gen PCSK9 aumentará la destrucción de los receptores, y elevará los niveles de colesterol; por el contrario, la pérdida de función del gen se traducirá en niveles extremadamente bajos de colesterol38, que fueron los que llevaron al descubrimiento de esta proproteína varios años después de que Goldstein y Brown39 ganaran el Premio Nobel de Medicina por el descubrimiento del RLDL40.
Alteración en el gen LDLRAP1Esta proteína permite la unión fácil del receptor con el ligando, la ApoB. La mutación de este gen disminuye la función y dificulta la formación del complejo RLDL-ApoB-LDL, con la elevación de los niveles de LDL como consecuencia.
Prevalencia de la hipercolesterolemia familiarSe estima que puede haber entre 14 y 34 millones de casos de HF en el mundo y que menos del 10% están diagnosticados y tan solo 5% de ellos son tratados de manera adecuada1,41,42. Históricamente, la prevalencia de la HFHe se ha estimado en un caso por cada 500 personas en la población general, y la de la HFHo en un caso por millón de habitantes; sin embargo, es una enfermedad subdiagnosticada y subtratada. De acuerdo con datos recientes, la prevalencia de la HFHe podría ser de un caso por 200 a 500 personas y de un caso por 300.000 a 600.000 personas para la HFHo42. Con estos datos, en Colombia se pueden estimar entre 96.000 y 240.000 y 160 y 300 las personas con HFHe y con HFHo, respectivamente. Es preciso tener en cuenta que hay grupos poblacionales en los que la prevalencia puede ser mayor por un efecto fundador o por consanguinidad. El efecto fundador consiste en la difusión por herencia de un gen en poblaciones con aislamiento geográfico o cultural. En poblaciones muy definidas, la prevalencia de HF es mucho mayor debido a este efecto43. A manera de ejemplo, en Sudáfrica se encontró una prevalencia de 1 caso de HFHe en 70 personas y un caso en 30.000 para HFHo44.
Diagnóstico fenotípico de la hipercolesterolemia familiarA pesar de ser una enfermedad genética, la HF es una condición tratable y controlable en la que es fundamental el diagnóstico correcto y precoz. En ocasiones éste no es preciso ya que no pasa del hallazgo de una dislipidemia16,45 y además no hay un código específico en la clasificación internacional de enfermedades (CIE 10) emitida por la Organización Mundial de la Salud (OMS) que facilite tener en mente esta entidad.
Características clínicasLa HF es una entidad que puede cursar asintomática u oligosintomática y cuyas manifestaciones derivan del depósito de colesterol en los órganos y particularmente de la aterosclerosis, que se revela principalmente como enfermedad coronaria46,47. Por este motivo, es importante diagnosticar la enfermedad en etapas tempranas, bien con un tamizaje dirigido o generalizado, o con búsqueda de casos que permitan controlar al paciente y al progenitor, quien posiblemente no conozca su nivel plasmático de cLDL48,49.
Niveles de lípidosEl patrón característico en la HF es un nivel de cLDL superior a 190mg/dL, con valores generalmente superiores a 500mg/dL en la HFHo. En algunos casos, puede existir una superposición de niveles de colesterol, que dificulta diferenciar clínicamente si se trata de HFHe o HFHo, aunque generalmente los valores son superiores en la HFHo. Los triglicéridos suelen ser normales; sin embargo, la hipertrigliceridemia no excluye el diagnóstico de HF si el resto de los criterios lo apoyan1–12,16. De otra parte, los niveles de cHDL pueden ser normales o discretamente bajos.
Enfermedad cardiovascularUna de las principales características de la HF es la enfermedad aterosclerótica cardiovascular prematura. Si no se trata adecuadamente la HFHe, el riesgo de enfermedad coronaria prematura es significativamente más alto en comparación con aquellos que no tienen la enfermedad. En el caso de los pacientes con HFHo no tratada, el riesgo de eventos cardiovasculares empieza antes de los 20 años y la muerte puede ocurrir antes de los 30 años. Son menos frecuentes las alteraciones en arterias carótidas, renales y femorales. Además, en la HFHo es característico el compromiso de la válvula aórtica y la aterosclerosis de la región supraaórtica, con la posible oclusión del ostium coronario que puede ocasionar la muerte súbita2–9,16.
Depósitos lipídicosLos xantomas tendinosos son patognomónicos de la HF. En los heterocigóticos se suelen observar entre la cuarta y la quinta décadas de la vida y están presentes en menos del 30% de los casos; la localización más frecuente es en los tendones de Aquiles y en los extensores de los dedos de la mano. En los casos de HFHo son especialmente notorios los xantomas cutáneos y tuberosos que pueden aparecer a partir del segundo año de vida, y son patognomónicos los xantomas interdigitales. En ocasiones, los xantomas se asocian con dolor articular o limitación funcional y a esto se agrega el efecto estético que muchas veces altera la calidad de vida. Algunos xantomas requieren resección quirúrgica; por ello, debe reforzarse entre los dermatólogos la necesidad de estudio y referencia de estos pacientes (figs. 3–5). El arco corneal no es patognomónico de la HF, pero ayuda a la sospecha diagnóstica cuando aparece antes de los 45 años (fig. 6).
Historia familiar



Es fundamental realizar un análisis familiar completo, que incluye el mayor número posible de generaciones tanto de forma ascendente como descendente a partir del caso índice. En casos de HF al menos uno de los progenitores suelen cursar con hipercolesterolemia significativa; en los casos poco frecuentes de hipercolesterolemia autosómica recesiva, los progenitores generalmente tienen niveles normales y sólo con la evaluación del árbol familiar o pedigrí se podrá encontrar el patrón recesivo50.
Tamizado. Cómo hacer la búsqueda activa de pacientes con hipercolesterolemia familiarLa búsqueda de pacientes con HF se hace principalmente con el perfil lipídico. Las estrategias dependen de la población seleccionada para la prueba.
Tamizado universalDetección de casos en la población general. Para el caso de interés, mediante la realización de perfil lipídico a todas las personas.
Tamizado selectivo o búsqueda de casosCuando hay antecedentes familiares de dislipidemia o enfermedad aterosclerótica prematura.
Tamizado en cascadaA partir de un caso de HF ya conocido (caso índice) se hace una búsqueda activa a los otros miembros de la familia.
Tamizado en cascada inversaLa búsqueda de casos en los familiares se hace de forma ascendente, en su origen genealógico.
En Colombia, la “Guía de práctica clínica de dislipidemia” propuso el inicio de tamizado universal en personas asintomáticas o sin factores de riesgo, a los 45 años tanto para hombres como para mujeres51; sin embargo, esta edad es tardía para diagnosticar la HF y prevenir el desarrollo de la enfermedad cardiovascular prematura. Por lo tanto, para el tamizado universal de HF se considera apropiado lo indicado por el National Heart, Lung, and Blood Institute y la American Academy of Pediatrics, que recomiendan un perfil lipídico entre los 9 y 11 años, que se repite entre los 17 y 21 años para efectos de confirmación9,52. En los casos de alto riesgo, el estudio debe iniciarse a los dos años.
Cómo sospechar un caso índice- -
Pacientes con niveles de cLDL mayores de 190mg/dL (sospecha de HFHe).
- -
Pacientes con niveles de cLDL mayores de 500mg/dL sin tratamiento o > 300mg/dL en personas con terapia farmacológica específica (sospecha de HFHo)9.
- -
Signos de depósito corporal de colesterol (xantomas)
- -
Historia personal o familiar de enfermedad cardiovascular prematura (hombres menores de 55 años o mujeres menores de 60 años).
- -
Historial familiar de hipercolesterolemia, con valores de cLDL en rango de HF.
- -
Si ambos padres tienen niveles de cLDL en rango de HFHe se debe sospechar HFHo.
- -
Cuando se encuentran niveles de cLDL inferiores a 300mg/dL, en pacientes tratados, no puede excluirse el diagnóstico de HFHo17.
Ante la sospecha de un caso índice se recomiendan acciones para la confirmación del diagnóstico y para iniciar el tratamiento. Las recomendaciones son:
- -
Realizar un análisis familiar para determinar el patrón de herencia.
- -
Descartar causas secundarias de dislipidemias como hipotiroidismo, diabetes, síndrome nefrótico o hepatopatías, entre otras.
- -
Aplicar los criterios de probabilidad validados para el diagnóstico clínico17–19.
- -
Idealmente medir los niveles de lipoproteína (a) (Lp(a) y de ApoB.
- -
Practicar una evaluación multidisciplinaria a cargo de profesionales con experiencia en dislipidemias severas que incluyan Nutrición, Psicología, Genética, Cardiología, Pediatría, Endocrinología, entre otras.
- -
Realizar estudios de aterosclerosis subclínica.
- -
Ordenar ecocardiograma transtorácico si hay soplos cardíacos.
- -
Efectuar estudios de confirmación genética si se considera necesario y está disponible.
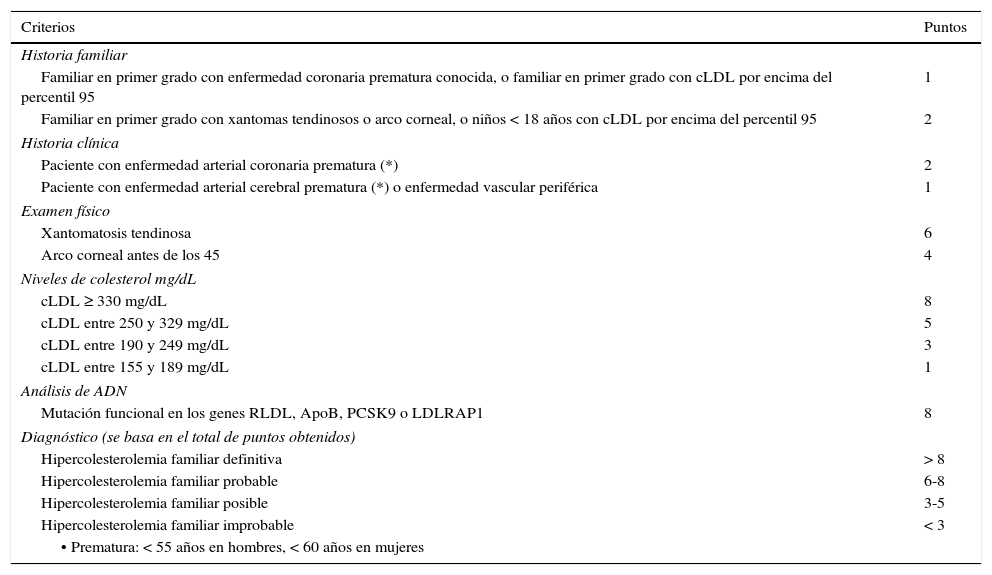
Existen una serie de criterios validados para el diagnóstico clínico en adultos, generalmente no recomendados en menores de 18 años: los criterios MEDPED (Make Early Diagnosis to Prevent Early Death)17, los del Simon Broome (Reino Unido) que sí permiten diagnosticar a menores de 18 años18 y los de la Red de clínicas de lípidos de Holanda (DLCN: Dutch Lipid Clinic Network)19. Se recomienda el uso de los criterios de la DLCN19 (tabla 1) ya que tienen validación genética. Para el diagnóstico genético, si ya se ha identificado una mutación causal en el caso índice no es necesario hacer secuenciaciones complejas en los familiares y es suficiente con un estudio dirigido a encontrar la mutación ya documentada en el caso índice. La evaluación con escalas de puntaje diagnóstico aplica para el caso índice y no para el tamizaje en cascada posterior17.
Red de Clínicas de Lípidos Holandesa (DLCN)19
| Criterios | Puntos |
|---|---|
| Historia familiar | |
| Familiar en primer grado con enfermedad coronaria prematura conocida, o familiar en primer grado con cLDL por encima del percentil 95 | 1 |
| Familiar en primer grado con xantomas tendinosos o arco corneal, o niños < 18 años con cLDL por encima del percentil 95 | 2 |
| Historia clínica | |
| Paciente con enfermedad arterial coronaria prematura (*) | 2 |
| Paciente con enfermedad arterial cerebral prematura (*) o enfermedad vascular periférica | 1 |
| Examen físico | |
| Xantomatosis tendinosa | 6 |
| Arco corneal antes de los 45 | 4 |
| Niveles de colesterol mg/dL | |
| cLDL ≥ 330 mg/dL | 8 |
| cLDL entre 250 y 329 mg/dL | 5 |
| cLDL entre 190 y 249 mg/dL | 3 |
| cLDL entre 155 y 189 mg/dL | 1 |
| Análisis de ADN | |
| Mutación funcional en los genes RLDL, ApoB, PCSK9 o LDLRAP1 | 8 |
| Diagnóstico (se basa en el total de puntos obtenidos) | |
| Hipercolesterolemia familiar definitiva | > 8 |
| Hipercolesterolemia familiar probable | 6-8 |
| Hipercolesterolemia familiar posible | 3-5 |
| Hipercolesterolemia familiar improbable | < 3 |
| • Prematura: < 55 años en hombres, < 60 años en mujeres | |
La HF es una enfermedad con predominio del patrón de herencia autosómico dominante, caracterizada por hipercolesterolemia desde el nacimiento y por un fenotipo que incluye xantomas y aterosclerosis prematura. Es preciso excluir causas secundarias de hipercolesterolemia y el diagnóstico diferencial debe hacerse con otras causas de hipercolesterolemia de base genética.
Hipercolesterolemia poligénicaEs la forma más común de hipercolesterolemia primaria, secundaria a la interacción entre numerosos genes y factores ambientales. La prevalencia en población general es aproximadamente del 4%. Los niveles de colesterol total suelen ser más bajos que en la HF (250-300mg/dL) y los triglicéridos son normales. Los antecedentes familiares de hipercolesterolemia son poco frecuentes (menos del 20% de los casos). La enfermedad coronaria suele aparecer después de los 50 años. No se asocia con xantomas pero sí con obesidad, diabetes, dieta inadecuada e hipertensión arterial.
Hiperlipidemia familiar combinadaEs un trastorno frecuente del metabolismo de los lípidos que afecta el 1 al 2% de la población general. Tiene un mecanismo de transmisión autosómico dominante como en la HF, pero hasta la fecha no se ha identificado un defecto molecular único asociado con este trastorno, razón por la cual no hay una prueba genética diagnóstica que la identifique. Se suele expresar a partir de la segunda década de la vida, y el 50% de los familiares del paciente pueden estar afectados. Se sospecha cuando en la familia hay varios individuos con trastornos lipídicos variables como cLDL mayor de 160mg/dL, hiperlipidemia mixta o hipertrigliceridemia con cHDL bajo y enfermedad coronaria prematura. No se asocia con xantomas, pero sí con mayor prevalencia de diabetes, hipertensión arterial, sobrepeso y obesidad.
Hiperlipoproteinemia tipo III (disbetalipoproteinemia)Es una enfermedad genética con patrón de herencia autosómico recesiva53, secundaria a alteraciones en el gen ApoE, ubicado en el cromosoma 19 (19q13.2), con predominio de la isoforma ApoE2. Su incidencia en la población general es de un caso en 10.000 personas55. Se caracteriza por la acumulación de residuos de lipoproteínas en el plasma y el desarrollo de aterosclerosis prematura; sin embargo, su fisiopatología es poco conocida y además de la presencia del genotipo E2E2 debe haber algún factor precipitante como diabetes mellitus, hipotiroidismo, obesidad, fármacos o daño renal54,56. La mayoría de los pacientes cursan con hipercolesterolemia e hipertrigliceridemia (usualmente mayor de 300mg/dL) y en algunos casos con xantomas estriados palmares, que son patognomónicos del trastorno. Algunos pacientes pueden ser normolipémicos e incluso hipolipémicos54,56. Se cree además que los hombres son más susceptibles, pues los estrógenos confieren un efecto protector a las mujeres56.
Xantomatosis cerebrotendinosaEn estos casos, el diagnóstico diferencial se da por la presencia de xantomas y no por los niveles de colesterol, que generalmente son normales. Es una enfermedad con patrón de herencia autosómico recesivo, secundaria a mutaciones en el gen CYP27A1, ubicado en el cromosoma 2 (2q35) y responsable de la síntesis de la enzima esterol 27 hidroxilasa57. La incidencia estimada de esta enfermedad es de 3 a 5 casos en 100.000 personas; sin embargo, en judíos marroquíes es de 1 en 10858. Se relaciona con depósitos de lípidos en el cerebro y en los tendones y se puede acompañar de diarrea crónica en la niñez, así como de aparición temprana de cataratas y problemas neurológicos en la edad adulta, como ataxia, convulsiones, demencia y depresión. Los hallazgos más comunes son: colesterol plasmático normal o ligeramente elevado, colestanol plasmático elevado, elevación urinaria de los alcoholes biliares 7 alfa-hidroxilados y deficiencia de la esterol 27-hidroxilasa59,60. Se han descrito casos en los que coexisten la HF y la xantomatosis cerebrotendinosa (XCT); en ellos, la presencia de HF parece proteger del daño neurológico propio de la XCT61.
SitosterolemiaEs una enfermedad con patrón de herencia autosómico recesivo, causada por mutaciones en los genes ABCG5 o ABCG8, ambos ubicados en el cromosoma 2 (2p21)62, que afecta los transportadores en el enterocito. Predomina en la edad pediátrica y es poco frecuente. Tiene características similares a la HFHo, como niveles elevados de colesterol (aunque no siempre) y xantomas. Los padres pueden cursar con niveles normales de colesterol. Las alteraciones de laboratorio más comunes incluyen niveles plasmáticos elevados de betasitosterol, campesterol, esigmasterol, colesterol e hiperapobetalipoproteinemia63,64. Estos pacientes pueden tener enfermedad coronaria prematura y responden satisfactoriamente al consumo estricto de fitoesteroles o a medicamentos como ezetimibe o secuestrantes de ácidos biliares.
Enfermedad aterosclerótica subclínica en la hipercolesterolemia familiarLos altos niveles plasmáticos de cLDL desde el nacimiento, en un individuo con HF, exponen a su sistema cardiovascular a una forma intensa y continua de partículas lipídicas y sustancias inflamatorias, que conduce a la formación temprana de placas ateroscleróticas65,66.
Ultrasonografía carotídeaPermite evaluar el incremento del espesor de la íntima-media (EI-M) de las arterias carótidas y la presencia de placas de ateroma. Se considera anormal un EI-M superior a 0,9mm. El método proporciona una medida de aterosclerosis subclínica y ha demostrado ser predictor de riesgo de eventos cardiovasculares67. El estudio ARIC mostró que la presencia de placa visible incrementa el riesgo cardiovascular68. En adultos se recomienda esta técnica por ser predictora de aterosclerosis generalizada69. La prueba no siempre ha sido aceptada para uso en niños70; sin embargo, en niños con HF se ha observado mayor EI-M antes de los 8 años en comparación con sus hermanos sin HF71. Se enfatiza que la técnica debe realizarse en centros con experiencia72.
Puntaje de calcio coronario y angiotomografía coronariaEl puntaje normal en unidades Agatston es cero, e implica ausencia de calcificación coronaria y pronóstico excelente. Un índice de calcio entre 100 y 399 se asocia con un incremento de cuatro veces el riesgo de muerte por enfermedad coronaria o infarto de miocardio mientras que un puntaje mayor de 400 se relaciona con un riesgo seis veces mayor en comparación con puntajes menores de 10073,74. En individuos asintomáticos, el puntaje de calcio coronario puede ser superior a la angiotomografía coronaria y al EI-M carotídeo para predicción de riesgo. El puntaje de calcio ha demostrado tener el mejor impacto en la reclasificación de riesgo entre todos los biomarcadores42,75,76. No se recomienda su uso en niños ni en adolescentes.
Angiorresonancia y resonancia magnéticaEn la actualidad, la sensibilidad y especificidad de esta técnica no son suficientes para recomendar el tamizado de la estenosis coronaria en personas asintomáticas en alto riesgo, como en los pacientes con HFHo. La resonancia magnética se reserva para evaluar la enfermedad ateromatosa de la aorta, la cual es frecuente y temprana en la HFHo, en la eventualidad de que la ecocardiografía aporte datos dudosos77,78.
Ecocardiografía transtorácicaEs útil en pacientes asintomáticos con HFHo y HFHe, principalmente en la HFHo, dada la acumulación temprana de colesterol y placas ateromatosas en la aorta ascendente, los ostia coronarios y el compromiso de las válvulas aórtica y mitral3, en especial cuando se detecta regurgitación valvular aórtica, que parece ser un marcador de inicio de compromiso aterosclerótico coronario79. De acuerdo con los hallazgos del ecocardiograma transtorácico y con el criterio clínico se realiza el ecocardiograma transesofágico80.
Ecocardiografía de estrés y perfusión miocárdicaSe recomienda si no se pueden practicar el EI-M carotídeo o el puntaje de calcio o cuando estas pruebas son positivas. En pacientes con HF es una recomendación clase IIb y nivel de evidencia C81.
Índice tobillo-brazoEs una prueba costo-efectiva para la detección de aterosclerosis subclínica. Un índice inferior a 0,9 indica una obstrucción mayor del 50% al flujo entre la aorta y las arterias distales de la pierna. Un índice entre 0,4 y 0,9 indica reducción moderada, en tanto que un resultado menor de 0,4 indica presencia de enfermedad vascular severa. Este índice detecta enfermedad obstructiva vascular en estados subclínicos, antes de la aparición de claudicación, y es buen predictor de enfermedad coronaria y cerebrovascular82.
Nuevos biomarcadoresLa medición de apolipoproteína B, el tamaño de las partículas y la densidad, la fosfolipasa A2 asociada con lipoproteína, la proteína C reactiva y otros biomarcadores séricos tienen poca aplicación en los pacientes con HF (Recomendación clase III, nivel de evidencia C)75,81. Recientemente se le ha dado importancia a la Lp(a) como factor de riesgo independiente del cLDL en la población con HF y se ha propuesto que deberían ser tratados sus niveles altos20.
Angiografía coronariaSolo se realiza si las pruebas de provocación de isquemia son positivas o en individuos con clínica de angina81.
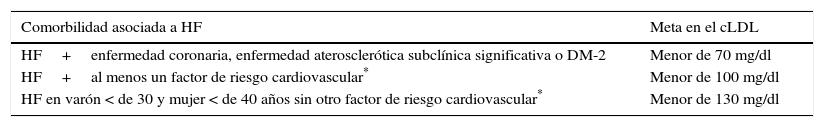
Metas del cLDL en pacientes con hipercolesterolemia familiarLos pacientes con HF deben ser considerados de alto riesgo cardiovascular; por ello, no se recomienda estratificar este riesgo con la ecuación de Framingham o el score europeo83–85. En la actualidad, las sociedades científicas internacionales plantean distintos objetivos en cLDL para los pacientes de alto riesgo cardiovascular. Así, la guía de práctica clínica basada en evidencia publicada en 2013 por la American Heart Association (AHA) y el American College of Cardiology (ACC)83 propuso como objetivo una reducción en cLDL superior al 50% en aquellos sujetos con cLDL mayor de 190mg/dL, donde se encuentran la mayoría de los pacientes con HF, y recomendó el uso de estatinas de alta efectividad y a las dosis más altas (atorvastatina, rosuvastatina)83. Por otra parte, la “Guía europea para el manejo de las dislipidemias”84 y la reciente “Guía de la Asociación Nacional de lípidos de los Estados Unidos de Norteamérica”85,86, proponen metas concretas en cLDL; específicamente, para pacientes con HF sugieren un valor menor de 100mg/dL. Otras recomendaciones internacionales están de acuerdo tanto con la reducción porcentual como con la consecución de metas en cLDL2,42. Por último, también se recomiendan metas en el cLDL de acuerdo con las comorbilidades asociadas con la HF2 (tabla 2).
Metas en el cLDL en HF
| Comorbilidad asociada a HF | Meta en el cLDL |
|---|---|
| HF+enfermedad coronaria, enfermedad aterosclerótica subclínica significativa o DM-2 | Menor de 70 mg/dl |
| HF+al menos un factor de riesgo cardiovascular* | Menor de 100 mg/dl |
| HF en varón < de 30 y mujer < de 40 años sin otro factor de riesgo cardiovascular* | Menor de 130 mg/dl |
Factor de riesgo cardiovascular en HF: varón > de 30 y mujer > de 40 años, tabaquismo, enfermedad coronaria prematura en familiares de primer grado, hipertensión arterial, cHDL < de 40mg/dL, Lp(a) > de 50mg/dL. Modificada de Mata P, Alonso R, Ruiz A, González-Juanatey JR, Badimón L, Díaz-Díaz JL, et al2.
Sin embargo, conseguir estas metas en la HF es difícil en muchos casos y constituye un reto para el personal médico a cargo, si se tiene en cuenta que los valores basales de cLDL suelen ser muy altos en la mayoría de individuos87,88. En el caso de Colombia, la “Guía colombiana de práctica clínica para la prevención, detección, diagnóstico, tratamiento y seguimiento de las dislipidemias en la población mayor de 18 años”51 propuso los mismos grupos de intervención que la guía del ACC/AHA (los pacientes con cLDL mayor de 190mg/dL se ubicarían en riesgo alto por considerarse que en este grupo estaría la mayoría de casos con HF)83.
Cambios terapéuticos en el estilo de vida en el tratamiento de la hipercolesterolemia familiarLa enfermedad cardiovascular prematura en pacientes con HF está determinada, además de los factores genéticos inherentes a la enfermedad, por factores ambientales y metabólicos adicionales que actúan en conjunto con la hipercolesterolemia42,65,89,90. Distintos estudios han demostrado el papel de factores de riesgo como el sexo masculino, la edad, el tabaquismo, el índice de masa corporal elevado, la hipertensión arterial, la diabetes, los niveles bajos de cHDL y los niveles altos de Lp(a) en el desarrollo y pronóstico de enfermedad cardiovascular en pacientes con HF20,89–92.
Todos los pacientes con HF deben recibir consejos acerca de los cambios terapéuticos en el estilo de vida y en todos los casos deberá advertirse sobre la importancia de modificar los factores de riesgo adicionales al cLDL elevado, además de enfatizar que la adopción de estas medidas no excluye ni sustituye el manejo farmacológico y que son fundamentales ya que tienen un efecto aditivo que facilita el logro de las metas terapéuticas. Igualmente, deberá recalcarse que la HF es un trastorno en cuyo tratamiento debe participar el núcleo familiar42,91,92. Los cambios terapéuticos en el estilo de vida deben estar a cargo de un equipo multidisciplinario y articulados con el personal de atención primaria en salud y se deben iniciar desde el diagnóstico42.
Cambios dietariosLas modificaciones dietarias para disminuir la ingesta de grasas saturadas, grasas trans y colesterol contribuyen a mejorar el perfil lipídico plasmático. En este sentido, una dieta tipo mediterránea, con frutas y verduras, baja en grasas saturadas y alta en grasas monoinsaturadas (aguacate, aceitunas, nueces y aceite de oliva), así como peces con alto contenido de ácidos grasos insaturados, puede tener efectos benéficos en personas con HF42,65. De acuerdo con las recomendaciones recientes de la Asociación Nacional de Lípidos de Norteamérica, hay controversia sobre si el consumo de colesterol aportado por un huevo al día se asocia con incremento del colesterol sérico y eventos cardiovasculares; parece existir una respuesta individual86. Puede recurrirse a suplementación dietaria con fitoesteroles y estanoles, que logran reducciones adicionales en los niveles de cLDL plasmáticos65. En el caso de niños y adolescentes, la alimentación podría constituir la base del tratamiento, toda vez que en ellos el recurso farmacológico puede ser más limitado que en los adultos; con estas medidas en este grupo etario podrían alcanzarse reducciones de hasta un 15% en los niveles de cLDL42,92. En estos pacientes es esencial un aporte adecuado de energía y nutrientes para permitir el crecimiento y desarrollo apropiados.
EjercicioEn todos los pacientes con HF se recomienda la práctica diaria de ejercicio aeróbico, que permita acumular al menos 150 minutos por semana. Esta recomendación debe ser reforzada en quienes tengan obesidad o sobrepeso, en cuyo caso se recomienda la práctica de ejercicio aeróbico durante 300 minutos a la semana. Antes de iniciar el régimen de ejercicio es pertinente en algunos pacientes realizar una valoración previa, con electrocardiograma, ecocardiograma o pruebas de provocación de isquemia según su pertinencia42.
Modificación de otros hábitos y factores de riesgoEs necesario moderar el consumo de alcohol, si existe el hábito, así como valorar el estrés psicosocial. Se debe evitar el hábito tabáquico y suspenderlo en los que ya lo tienen. También deberá desaconsejarse el tabaquismo pasivo en las familias con HF65. Los factores de riesgo adicionales (diabetes mellitus, hipertensión arterial) deben controlarse de manera más estricta en quienes tienen HF por su alto nivel de riesgo basal. El médico y el equipo de salud deben tener claro que aún con adherencia estricta a los cambios terapéuticos en el estilo de vida, estos pueden tener poco impacto en la reducción del cLDL (entre el 10 y 15%), y aún menos en HFHo, concepto que debe explicársele al paciente y a su familia, para no dar lugar a falsas expectativas, causa frecuente de suspensión de las medidas.
Tratamiento tradicional para la hipercolesterolemia familiarEn la HF, el uso de los hipolipemiantes tradicionales necesariamente debe ir asociado con los cambios terapéuticos en el estilo de vida y con el tratamiento simultáneo de los otros factores de riesgo que estén presentes. El mensaje principal es el inicio del tratamiento una vez se ha hecho el diagnóstico.
EstatinasHan demostrado beneficio cardiovascular al reducir la morbilidad y la mortalidad total y la de origen cardiovascular en población de alto riesgo cardiovascular; así mismo, han demostrado eficacia en reducir los niveles de colesterol. El beneficio de las estatinas en pacientes con HFHe también se ha demostrado en estudios observacionales en pacientes con y sin cardiopatía isquémica93,94. Aunque los pacientes con HF han sido excluidos de los grandes estudios de intervención para demostrar beneficio cardiovascular, es probable que en los estudios 4S, WOSCOPS y LRC-CPPT se hayan incluido casos con HF de acuerdo con los niveles de cLDL mayores de 190mg/dL que tenían parte de los pacientes. De manera interesante, también en este subgrupo se demostró reducción de eventos cardiovasculares95–97. De igual forma, en los pacientes con HFHo se ha observado cierta respuesta al tratamiento con estatinas a pesar de tener poca o nula actividad del receptor LDL98–100. En este punto, en la era previa a las estatinas, la sobrevida de los pacientes con HFHo era alrededor de los 13 años, pero hoy su uso la ha ampliado a 33 años100. Los pacientes con HF difícilmente logran las metas estipuladas, hecho que se explica por la gran elevación plasmática de los niveles de colesterol y por la menor respuesta a las estatinas en esta población, lo cual conlleva riesgo cardiovascular residual y requerimiento frecuente de combinación de hipolipemiantes. Las estatinas más efectivas como la atorvastatina y la rosuvastatina en sus dosis más altas, reducen el cLDL plasmático entre un 50 y un 58%; en los individuos con HF estos porcentajes son insuficientes para alcanzar las metas en el cLDL, sin embargo, son el pilar fundamental en el tratamiento y la base para los diferentes esquemas de combinación.
EzetimibeEs un inhibidor selectivo de la absorción intestinal del colesterol; inactiva de forma reversible la acción de la proteína transportadora intestinal similar a la proteína 1 Niemann-Pick C1 (NPC1L1). Tiene una vía metabólica hepática diferente de la del citocromo P450, que brinda la posibilidad de menor interacción farmacológica101. Su efecto sinérgico con las estatinas reduce los valores plasmáticos del cLDL. Se ha demostrado su eficacia en el grupo de pacientes con HF; ofrece una reducción adicional alrededor del 20% en el cLDL102–104. El espacio principal para su uso es el grupo de pacientes que no llegan a metas en el cLDL a pesar de la dosis máxima de las estatinas más efectivas. Su combinación con estatinas tiene buena tolerancia y baja probabilidad de efectos adversos. También ha demostrado ser seguro en la población pediátrica104.
Secuestrantes de ácidos biliaresDisminuyen la recaptación de grasas en la circulación enterohepática y aumentan la eliminación de sales biliares. No tienen absorción sistémica, lo que explica los mínimos efectos adversos generales; la mayoría de los síntomas se limitan al sistema digestivo105,106. Su inconveniente es la administración durante varias veces al día y la frecuente intolerancia por molestias digestivas (estreñimiento, náuseas, dispepsia, vómito y flatulencia), que los hacen de pobre adherencia. La reducción promedio de los niveles plasmáticos de cLDL es de 15 a 20mg/dL. Pueden empeorar la hipertrigliceridemia. En nuestro medio se encuentra disponible la colestiramina en sobres de 4 gramos para disolver en agua y tomar 3-4 veces al día. En los pacientes con HF la mayor evidencia la tiene colesevelam, que además presenta menos efectos adversos107.
Niacina y fibratosLa niacina puede reducir los niveles plasmáticos del cLDL aproximadamente en 20%. Sus inconvenientes principales son los efectos adversos (rubor, prurito y náuseas) y la no demostración de reducción de eventos cardiovasculares, al menos en pacientes con enfermedad coronaria86. Los fibratos en la HF tienen poca utilidad86.
Utilidad de nuevas moléculas hipolipemiantes: lomitapide, mipomersén y anticuerpos monoclonales antiPCSK9 en hipercolesterolemia familiarLa mayoría de pacientes con HF no consiguen el objetivo terapéutico en el cLDL mencionado anteriormente191. Recientemente, distintas agencias regulatorias ha probado nuevos fármacos hipolipemiantes que buscan reducir aún más los niveles de cLDL en especial en la población con HFHo, HFHe o con cLDL muy elevado, de difícil tratamiento.
Fármacos que alteran la producción y secreción de lipoproteínas con ApoB (lomitapide y mipomersén)LomitapideEsta molécula de administración oral inhibe la proteína de transferencia microsomal de triglicéridos (MTTP, del inglés microsomal triglyceride transfer protein), una enzima clave en el ensamblaje y la secreción de lipoproteínas que contienen ApoB, tanto en el hígado (VLDL con ApoB 100) como en el intestino (quilomicrones con ApoB 48). Por tanto, la inhibición de esta enzima disminuye la síntesis y secreción de la VLDL y de los quilomicrones y así es capaz de reducir los niveles plasmáticos de CT, TG, cLDL y VLDL. Debido a su mecanismo de acción, el bloqueo de la MTTP no tiene efecto en los receptores de LDL, cualidad que lo hace muy atractivo para el manejo de la HFHo108–110. El lomitapide ha demostrado ser útil como complemento a una dieta baja en grasa y otros tratamientos reductores del colesterol, incluyendo la aféresis de LDL cuando sea posible, para bajar el cLDL en un 40% en pacientes con HFHo. De acuerdo con las recomendaciones de la EMA y la FDA se sugiere comenzar con una dosis de 5mg por vía oral una vez al día (2 horas después de la cena) y titular cada 4 semanas según tolerancia, hasta llegar a una dosis máxima de 60mg/día. Además, se recomienda realizar una dieta estricta con menos del 20% de aporte calórico en grasas, con suplementación de vitamina E y ácidos grasos esenciales. Se hace mención especial en no utilizar dosis altas de estatinas de manera concomitante (por ejemplo, dosis de simvastatina mayor de 40mg diarios). Así mismo, debe realizarse periódicamente un análisis de los niveles de transaminasas (ASAT y ALAT) y buscar de manera intencionada la presencia de esteatosis hepática.
MipomersénEs un oligonucleótico antisentido que se une a la secuencia del ARN mensajero que codifica la ApoB 100, inhibe su traducción ribosomal y evita así la formación de la proteína, fenómeno que lleva a la disminución en el ensamblaje de VLDL. Fue aprobado por la FDA en enero de 2013 para la HFHo como terapia adicional del tratamiento habitual. Se aplica semanalmente en forma subcutánea y reduce los niveles plasmáticos de cLDL entre 25 y 36%, de TG, de Lp(a), de ApoB y de colesterol no-HDL111–118. Las lipoproteínas que contienen ApoB 48, como los quilomicrones, no se ven afectadas porque su origen es intestinal y no hepático119,120. El efecto adverso más frecuente, y que está relacionado con su mecanismo de acción, es la esteatosis hepática. También pueden aparecer reacciones en el sitio de inyección. Se describe la aparición de síntomas similares a un cuadro de influenza en el 70% de los pacientes, pero estos síntomas disminuyen en el mediano plazo. No está disponible en Colombia.
Fármacos que intervienen en la inhibición de la proproteína convertasa de subtilisina/kexina tipo 9 (PCSK9)La PCSK9 es una proteína enzimática de la familia de la subtisilina de serinas-proteasas, que se sintetiza primariamente en el hígado, aunque también se encuentra en el intestino y riñón. La PCSK9 produce una subregulación de la expresión del complejo RLDL-LDL; dicho complejo es internalizado y degradado en los lisosomas, lo que resulta en degradación del RLDL; como resultado, se reduce la recirculación de RLDL a la membrana celular y por lo tanto menor captación de cLDL y finalmente aumento de sus niveles plasmáticos. La inhibición de la síntesis de PCSK9, el antagonismo de su interacción con el RLDL o el incremento de su depuración son estrategias que pueden modificar el nivel de cLDL. En la actualidad se cuenta con medicamentos que hacen su efecto por los dos primeros mecanismos (inhibición de la síntesis de PCSK9 y por el antagonismo de la interacción con el RLDL). Las terapias biológicas con anticuerpos monoclonales (ACm) inhiben la interacción entre la PCSK9 y el RLDL, lo cual permite el reciclaje normal del RLDL. En la actualidad hay tres anti-PCSK9 (ACm/PCSK9): alirocumab, evolocumab y bococizumab, de los cuales los dos primeros tienen ya aprobación clínica por parte de la FDA y de la EMA; el tercero se encuentra en estudios de fase III. Se han estudiado sus efectos en monoterapia y adicionados a estatinas o a otros hipolipemiantes, en pacientes con riesgo cardiovascular alto en personas con intolerancia a las estatinas y en pacientes con HFHe e HFHo121,122.
AlirocumabEste ACm/PCSK9 se administra por vía subcutánea. Fue aprobado por la FDA en julio de 2015 y por la EMA en septiembre del mismo año, para pacientes con HFHe como adición a la terapia con la máxima dosis tolerada de estatinas y dieta, en pacientes con enfermedad cardiovascular aterosclerótica que requieran reducción adicional de cLDL o en intolerantes a las estatinas. El programa de estudios clínicos ODYSSEY comprende 14 estudios clínicos experimentales, con duración entre 24 y 104 semanas y más de 23.500 pacientes de diferentes poblaciones, en riesgo cardiovascular. Los estudios muestran reducción del cLDL y bajo porcentaje de eventos adversos. Se ha utilizado en monoterapia o en terapia asociada con estatinas o ezetimibe, en pacientes con alto riesgo para enfermedad cardiovascular, en intolerantes a las estatinas y en HFHe. Con la aplicación subcutánea de 150mg cada 15 días, el promedio de reducción del cLDL es del 50%. La respuesta también es favorable para el colesterol total, ApoB, colesterol no-HDL y Lp(a)123–128. El estudio ODYSSEY long-term, demostró, en 2.341 pacientes de alto riesgo para eventos cardiovasculares, una efectividad en la reducción del cLDL del 62% con alta seguridad129. A la fecha, se evalúa esta molécula en cuanto a resultados cardiovasculares a largo plazo (estudio ODYSSEY outcomes).
EvolocumabEste ACm/PCSK9, también para administración subcutánea, fue aprobado en mayo de 2015 por la EMA y en agosto del mismo año por la FDA para el tratamiento de adultos con HFHe, mayores de 12 años con HFHo que no logran alcanzar las metas en el cLDL con las dosis máximas de estatinas tolerables y dieta, y pacientes con enfermedad cardiovascular aterosclerótica que requieren reducción adicional en el cLDL o que son intolerantes a las estatinas. Los estudios muestran un efecto seguro en la reducción del cLDL tanto en monoterapia como en terapia asociada con estatinas o ezetimibe. Al adicionarse a las estatinas, el evolocumab logra una reducción promedio del cLDL del 51%; además, se asocia con una reducción sustancial en el nivel de colesterol total, ApoB, colesterol no-HDL y Lp(a). La dosis de 420mg administrada cada 4 semanas tiene una eficacia similar a 140mg cada 2 semanas, pero con la dosificación mensual se observa mayor variabilidad en los niveles de cLDL debido al consumo de los ACm que tienden a presentarse al final de las cuatro semanas130. El programa de estudios clínicos fase III, PROFICIO, consta de 22 experimentos clínicos y planea incluir aproximadamente 35.000 pacientes para evaluar el evolocumab en diferentes poblaciones con riesgo cardiovascular131–137. Recientemente se publicó el estudio OSLER, el cual también demostró en 4.465 pacientes una eficacia para reducir el cLDL en un 61% y alta seguridad138. En la actualidad se realiza el estudio FOURIER sobre eventos cardiovasculares a largo plazo.
BococizumabTres experimentos clínicos de fase III (SPIRE) avalúan la seguridad y eficacia del bococizumab sobre el cLDL en personas de riesgo cardiovascular alto y muy alto que reciben simultáneamente manejo con estatinas y ezetimibe130,139.
En los tres ACm/PSCK9 los estudios en fases I, II y III han mostrado que dichas moléculas son bien toleradas y no se han producido efectos adversos serios. No se espera que haya interacciones con las estatinas u otros fármacos ya que los ACm/PCSK9 no se metabolizan por el citocromo P450. En personas con intolerancia a las estatinas se produjeron mialgias hasta en un 15%, aunque no se relacionaron con la dosis del medicamento. Se han reportado hasta la fecha casos esporádicos de prurito, aumento en los niveles de CPK y vasculitis. La reducción del cLDL con evolocumab y alirocumab es en promedio de 50 a 60mg/dL respecto a los niveles basales140,141. Estas moléculas a la fecha no han sido aprobadas para su uso en Colombia.
Aféresis de LDL, trasplante hepático y otros enfoques terapéuticos en hipercolesterolemia familiarAféresis de LDLEs una opción terapéutica para los pacientes con HFHo que no logran reducir su cLDL de manera satisfactoria con los hipolipemiantes disponibles. Ha demostrado beneficios para la prevención de eventos cardiovasculares y en los pacientes con HF y aterosclerosis aórtica y coronaria142. Es una estrategia terapéutica costo-efectiva y segura. Se han descrito casos de HFHo tratados durante más de veinte años, en los que se ha demostrado la eliminación de xantomas, la regresión angiográfica de las lesiones ateromatosas coronarias y la reducción de episodios coronarios mortales y no mortales143–146. Existen diferentes técnicas de aféresis: plasmaféresis de doble filtración, inmunoabsorción específica, absorción mediante sulfato dextrano, precipitación extracorpórea de la LDL o absorción directa de lipoproteínas (DALI), que permiten la posibilidad de extraer las partículas de LDL a partir de sangre entera en lugar de hacerlo a partir del plasma como ocurre en los cuatro primeros métodos. La reducción de cLDL y Lp(a) conseguida está entre 50 y 75% de los niveles basales y se pueden obtener niveles de cLDL cercanos a los normales cuando se realiza semanalmente o cada dos semanas144. Está indicada en las siguientes situaciones: 1) HFHo a partir de los 5 años y siempre antes de los 8 años (si se requiere), 2) HF con enfermedad coronaria sintomática y cLDL mayor de 200mg/dL, a pesar de tratamiento farmacológico intenso, 3) HF con enfermedad coronaria progresiva sin posibilidad de revascularización, 4) cLDL mayor de 125mg/dL y Lp(a) mayor de 60mg/dL, a pesar de tratamiento farmacológico intenso, 5) En el embarazo y la lactancia durante el tiempo de suspensión del tratamiento farmacológico142–152. La edad de inicio, la frecuencia y la duración del tratamiento dependen de la disponibilidad, los costos, la necesidad clínica de lograr el objetivo de cLDL según el riesgo cardiovascular, la severidad de la enfermedad y la elección del paciente. La aféresis de LDL es un procedimiento bien tolerado, con efectos adversos en menos del 5%, que incluyen hipotensión, dolor abdominal, náuseas, hipocalcemia, anemia ferropénica y aquellos relacionados con el acceso venoso153. A la fecha no está disponible en Colombia.
Trasplante hepático y otras intervenciones quirúrgicasSe considera en los pacientes con HFHo cuando el resto de los tratamientos utilizados fallan, y en este caso se remiten a un centro con alta experiencia. La reducción de cLDL que se logra con el trasplante es equivalente a la que se obtiene con la combinación de estilo de vida, fármacos y aféresis de LDL. Otras razones para su uso limitado son el alto riesgo de complicaciones trans- y postoperatorias, mortalidad, escasez de donantes y necesidad de tratamiento inmunosupresor de por vida154.
La cirugía de puente ileal parcial o porto-cava no son procedimientos recomendados a la fecha155,156.
Tratamiento de la hipercolesterolemia familiar en la niñez y la adolescenciaEn niños y adolescentes con HF se ha demostrado la presencia de lesiones ateroscleróticas tempranas, secundarias a la hipercolesterolemia propia de esta entidad y en algunos casos agravada por la coexistencia con otros factores de riesgo157. En ocasiones, desde temprana edad hay anormalidades en el EI-M carotídeo y en el puntaje de calcio158–160. Tanto la HFHe como la HFHo son la principal causa de enfermedad cardiovascular prematura en pediatría161, razón por la cual se justifican las medidas que permitan el diagnóstico oportuno y el abordaje terapéutico temprano y óptimo157–162.
Recomendaciones generalesLa tabla 3 esquematiza las recomendaciones generales para niños con HF o sospecha de HF163.
Recomendaciones generales para niños con HF
| En los pacientes con perfil lipídico alterado se debe completar una evaluación clínica. Se indagará sobre los antecedentes cardiovasculares, el perfil lipídico de los familiares en primer grado (padres, hermanos) y la condición de riesgo cardiovascular del paciente. |
| Si el paciente presenta obesidad o sobrepeso y se sospecha dislipidemia secundaria al peso, el tratamiento debe dirigirse a normalizarlo. |
| Si el cLDL es mayor de 190 mg/dL, confirmado con segunda medición, el niño deberá derivarse al equipo o pediatra especialista en lípidos. |
| Es probable que un niño con cLDL mayor de 150-160 mg/dL y con uno de sus padres con hipercolesterolemia, tenga HF. En este caso, se inicia dieta hipolipemiante y se evalúa la respuesta a los 3-6 meses. Si persiste el cLDL mayor de 150 mg/dL se deriva al equipo especializado. |
| Los niños con cLDL mayor de 150-160 mg/dL y condiciones de riesgo cardiovascular o antecedente familiar de enfermedad cardiovascular prematura, deben referirse al equipo especializado. |
| Los padres con perfil lipídico alterado deberán derivarse al especialista respectivo. |
Adaptado del Consenso sobre manejo de las dislipidemias en Pediatría163.
Las escalas de probabilidad de certeza clínica17–19 no fueron diseñadas para niños; por ello en esta población la aproximación diagnóstica de HF se hace mediante un cLDL mayor de 190mg/dL o mayor de 150mg/dL con confirmación genética o evidencia de transmisión vertical o enfermedad coronaria prematura en sus progenitores166. La HFHo es una enfermedad que suele manifestarse en la infancia; su diagnóstico se hace cuando el cLDL es superior a 500mg/dL sin tratamiento o mayor de 300mg/dL con tratamiento, presencia de xantomas antes de los 10 años, historia de hipercolesterolemia en ambos padres o diagnóstico genético167.
Edad de detecciónSe considera apropiado tamizar con perfil lipídico entre los 9 y 11 años. En los casos de alto riesgo el estudio debe iniciarse a los dos años9,52.
Metas de cLDLSe propone un valor menor de 130mg/dL en los mayores de 14 años y menor de 160mg/dL en menores de 14 años sin otros factores de riesgo2. Es preciso considerar metas más estrictas ante la presencia de otros factores de riesgo o de historia familiar de enfermedad cardiovascular prematura. También se propone que en niños entre 8 y 10 años se reduzca el cLDL al menos en un 50% del valor basal, en tanto que en mayores de 10 años se recomienda conseguir una meta en cLDL de menos de 130mg/dL7. Se debe tener en consideración la dificultad para lograr estas metas o valores aceptables de cLDL, principalmente en HFHo164,165.
Tratamiento no farmacológicoPara niños con perfil lipídico normal pero con antecedente familiar para riesgo cardiovascular se diseñaron las guías dietarias CHILD-1 (Cardiovascular Health Integrated Lifestyle Diet) y para los niños con hipercolesterolemia se dispone de la CHILD-2, que intensifica la restricciones en la grasa dietaria157; ambas están disponibles en la página web del Instituto Nacional del Corazón, Pulmón y Sangre de los Estados Unidos (www.nhlbi.nih.gov/health-pro/guidelines).
Tratamiento farmacológicoEstatinasPara niños entre los ocho y los diez años solo la pravastatina ha sido aprobada por la FDA y por la EMA168. Tanto pravastatina, lovastatina y simvastatina como atorvastatina, están aprobadas por la FDA para mayores de 10 años8. La rosuvastatina está aprobada por la FDA y por la EMA en HF en mayores de 10 años168. En niños, el tratamiento con estatinas se ha considerado eficiente y seguro a corto plazo pero los efectos con su uso prolongado aún se desconocen170,171. Las limitantes para su utilización antes de los 8 años se relacionan con el desarrollo cerebral, puesto que se considera al colesterol un componente esencial de la mielina172; sin embargo, en los niños con HFHo se pueden usar a partir del segundo año de vida, teniendo en cuenta que no hay evidencia de paso de colesterol de la sangre al sistema nervioso central y se ha demostrado que el cerebro lo sintetiza de novo173. Las estatinas disponibles pueden inhibir en forma parcial la síntesis de colesterol cerebral, lo que significaría un potencial riesgo de daño en niños pequeños174.
FibratosSu seguridad y eficacia no está establecida en pediatría165,171.
EzetimibeLa dosis de 10mg al día asociada con estatinas logra una reducción adicional en el cLDL cercana al 20%175,176. No está establecida su eficacia y seguridad en menores de 10 años.
ColestiraminaSe recomienda como segunda opción para buscar metas o cuando hay contraindicación o intolerancia a las estatinas. No tiene efectos sistémicos y puede reducir el cLDL entre un 10 y un 20%177. Se inicia a dosis de dos gramos al día y se incrementa hasta cuatro gramos al día, repartida en tres a cuatro dosis. La adherencia es baja por sus efectos gastrointestinales. Se recomienda usar suplementos de vitaminas liposolubles.
Aféresis de LDL y trasplante hepáticoYa mencionados previamente.
En la figura 7 se propone un algoritmo sobre el tratamiento en la población pediátrica y adolescente2,9.

Algoritmo de tratamiento en niños y adolescentes con hipercolesterolemia familiar.
* Más eficaz a mayor dosis, dependiendo de la tolerancia.
** Inicio tan temprano como sea posible, no más tarde de los dos años, cada una o dos semana.
ξ Según tolerancia.
+ Aprobado por la FDA en adultos con HFHo. Aprobado por la EMA.
++ Aprobado por la FDA en adultos con HFHo.
Adaptado de: Diagnóstico y tratamiento de la hipercolesterolemia familiar en España2 y Consenso en hipercolesterolemia familiar de la Sociedad europea de aterosclerosis9.
Las principales causas secundarias de hipertrigliceridemia y de cLDL elevado en la mujer, incluyen el uso de estrógenos, anticonceptivos, embarazo y menopausia; por ello se analizan estos aspectos en personas con HF.
AnticoncepciónLas mujeres con HF en edad reproductiva deben recibir consejería gestacional y recomendaciones sobre anticoncepción e instrucciones para suspender la estatina, el ezetimibe, los fibratos o la niacina, desde uno a tres meses antes de suspender los métodos de planificación familiar178–181. Los métodos de anticoncepción de barrera, dispositivos intrauterinos, ligadura de trompas o vasectomía se prefieren sobre los anticonceptivos orales (ACO) porque no afectan el perfil lipídico ni el riesgo cardiovascular. En caso de utilizar ACO se opta por aquellos con bajo contenido de estrógenos181.
EmbarazoDurante el embarazo, los niveles de colesterol aumentan entre 30 y 50% (hasta 66% el cLDL) como resultado del incremento en la síntesis hepática de colesterol secundario a los niveles elevados de estrógenos, siendo mayor el incremento en las mujeres con HF182–185. Este aumento de colesterol empieza a partir del primer trimestre y se acentúa en el tercero; los niveles de triglicéridos se elevan hasta tres veces182. La hipercolesterolemia y la hipertrigliceridemia gestacionales tienen explicación fisiológica por la necesidad de incrementar la síntesis de esteroides sexuales y mantener un adecuado aporte de nutrientes para la madre y el feto.
Consecuencias de la hipercolesterolemia en el embarazoNo se ha definido aun si el aumento de los niveles de colesterol durante el embarazo en pacientes con HF se asocia con mayor aterosclerosis o riesgo cardiovascular. Se ha descrito mayor riesgo de parto prematuro, bajo peso al nacer y abortos recurrentes150,182,185. El único medicamento hipolipemiante que se puede administrar durante el embarazo son los secuestrantes de ácidos biliares (colestiramina, colesevelam), pero deben considerarse sus efectos adversos (estreñimiento, dispepsia y aumento de triglicéridos), que son más frecuentes durante el tercer trimestre del embarazo, los cuales se deben balancear con la reducción del cLDL que es del 15-20%. Si se utilizan estos medicamentos, se recomienda emplear suplementos de las vitaminas liposolubles. Otra opción, dependiendo de la condición y riesgo cardiovascular del paciente, es la aféresis de cLDL9,150,178,181–183,186,187.
Consecuencias del colesterol en el fetoHay controversia sobre si el feto adquiere y utiliza el colesterol materno para sus requerimientos metabólicos; se ha descrito que la hiperlipidemia durante el embarazo se asocia con aterosclerosis en las arterias espirales uteroplacentarias, hipercoagulación, trombosis, infarto placentario e insuficiencia placentaria, que pueden llevar a complicaciones fetales150,184,185. Adicionalmente, se han identificado cambios en la aorta fetal que determinan una mayor susceptibilidad a largo plazo en los niños para desarrollar estrías grasas y aterosclerosis. Se han encontrado lesiones aórticas preateroscleróticas más abundantes y más largas, así como mayor progresión de estas en comparación con los hijos de madres no hipercolesterolémicas182,188.
Resumen de puntos principales en la hipercolesterolemia familiarSe extractan por medio de preguntas, los principales aspectos para tener en cuenta en HF.
¿Qué es la HF?Es una alteración genética presente desde el nacimiento, caracterizada por niveles plasmáticos marcadamente elevados de cLDL y por enfermedad coronaria prematura1–12.
¿Cuántos tipos de mutaciones intervienen?De acuerdo con la mutación en sus genes, hay cuatro tipos. Todos tienen en común la elevación plasmática del cLDL:
- a)
Mutaciones en el gen que codifica el receptor del LDL (RLDL)26.
- b)
Mutación en el gen que codifica la apolipoproteína B (ApoB)27.
- c)
Mutaciones en el gen que codifica la proproteína convertasa de subtilisina kexina 9 (PCSK9) y que expresan ganacia en su función29.
- d)
Mutaciones en el gen que codifica la proteína adaptadora del receptor del LDL tipo 1 (LDLRAP1)30.
- a)
HFHe: es la forma más común, en la cual el paciente está afectado por mutaciones solamente en un alelo, heredado de uno de sus progenitores.
- b)
HFHo: el descendiente está afectado por mutaciones en dos alelos idénticos, heredados de sus dos progenitores. Se ha clasificado convencionalmente como HFHo con RLDL negativo o nulo, en quienes tienen actividad del RLDL menor del 2% e HFHo con RLDL defectuoso en quienes tienen actividad del RLDL entre el 2 y el 25%.
- c)
Heterocigóticos compuestos: ambos alelos tienen mutaciones diferentes9,13.
- d)
Heterocigóticos dobles: presentan mutaciones distintas en dos genes diferentes; uno de ellos es el gen del RLDL y la otra mutación puede ser ApoB o PCSK99,13.
La prevalencia histórica de la HFHe es de 1 en 500 habitantes y la de la HFHo de 1 en 1 millón de habitantes. Sin embargo, debe ser más alta puesto que la HF está subdiagnosticada y además se han demostrado prevalencias más altas que las mencionadas en algunas poblaciones16.
¿Cuáles son los niveles plasmáticos de cLDL en pacientes con HF?En general los pacientes adultos con HFHe tienen niveles plasmáticos de cLDL superiores a 190mg/dL y los niños y adolescentes niveles superiores a 160mg/dL. Los afectados con HFHo muestran niveles de cLDL por encima de 500mg/dL sin tratamiento farmacológico y mayores de 300mg/dL con tratamiento. Se ha demostrado una gran variabilidad en los niveles de cLDL y el fenotipo de HF3,9. En algunos casos puede ser difícil predecir si se trata de HFHe o HFHo solo con los niveles plasmáticos de cLDL. Es importante recordar que pueden encontrarse concentraciones anormales de cLDL sin que haya HF.
¿Cuáles son las consecuencias de la HF?Los altos niveles plasmáticos de cLDL en la HF llevan a aterosclerosis prematura, principalmente de las arterias coronarias14,15. Aproximadamente 20% de los infartos de miocardio antes de los 45 años y 5% antes de los 60 pueden ser atribuidos a HF; a los 50 años, los hombres y las mujeres con HF tienen un riesgo de evento cardiovascular del 50 y el 30%, respectivamente16. La HF también induce acumulación de colesterol en la piel, en la córnea, en los senos coronarios o en la válvula aórtica, condiciones más comunes, más precoces y más severas en la HFHo3,9.
¿Cómo se diagnostica la HF?Para diagnosticar la HF se recomienda analizar cinco criterios:
- 1)
Niveles plasmáticos de cLDL.
- 2)
Signos clínicos.
- 3)
Historia familiar de enfermedad coronaria prematura o de niveles altos de cLDL.
- 4)
Presencia de otras causas de elevación del cLDL.
- 5)
Estudio genético.
Se han propuesto tres guías de criterios clínicos para hacer el diagnóstico en adultos, aunque la Guía holandesa es la más utilizada17–19. En niños el diagnóstico se sospecha con cLDL mayor de 190mg/dL o mayor de 150mg/dL con confirmación genética de HF o evidencia de transmisión vertical o enfermedad coronaria prematura169. Para el diagnóstico de HFHo se incluyen las siguientes variables: a) Confirmación genética o b) cLDL > 500mg/dL sin tratamiento y > 300mg/dL con tratamiento, junto con c) Xantomas cutáneos o tendinosos antes de los 10 años o d) Niveles de cLDL sin tratamiento en ambos progenitores compatibles con HFHe167.
¿Cuáles son las metas para el cLDL en la HF?No hay consenso sobre cuáles deben ser las metas en la HF. La Guía americana de ACC/AHA83 y la colombiana51 especifican que no hallaron evidencia para el concepto de metas para el cLDL y aceptan que un nivel de cLDL mayor de 190mg/dL (donde estarían las personas con HF) equivale a riesgo alto, y recomiendan reducir el cLDL en más del 50% utilizando estatinas de alta intensidad a las dosis más altas. Es difícil lograr estas reducciones con la terapia mencionada, especialmente en personas con HFHo87; por ello se recomienda solicitar perfil lipídico a los dos meses de iniciada la estatina y de acuerdo con su resultado, buscar valores aceptables en el cLDL asociando otros hipolipemiantes. Otra propuesta en HF es buscar las siguientes metas en el cLDL2: 1) cLDL menor de 70mg/dl en pacientes con enfermedad aterosclerótica, diabetes mellitus tipo 2, enfermedad subclínica importante, diabetes mellitus tipo 1 con compromiso de órgano blanco, 2) cLDL menor de 100mg/dl en presencia de factores de riesgo cardiovascular, 3) cLDL menor de 130mg/dl en niños o adultos y en ausencia de factores de riesgo diferentes a HF2. Se deben considerar metas más estrictas ante la presencia de otros factores de riesgo o de historia familiar de enfermedad cardiovascular prematura. Otra propuesta emitida en 2015 y de origen europeo, recomienda que en niños entre 8 y 10 años el cLDL se reduzca en 50% de los niveles iniciales y en mayores de 10, la meta sea de menos de 130mg/dL7.
¿Cuál es el tratamiento de la HF?Los CTEV son los pilares del tratamiento, pero son insuficientes para lograr niveles óptimos de cLDL y por ello deben estar asociados con la terapia hipolipemiante2,8,12,21. El tratamiento farmacológico se inicia con las estatinas más efectivas y a las dosis más altas51,83. En un alto porcentaje de los tratados con estas condiciones se logra reducir el riesgo de eventos coronarios94. Con frecuencia se requiere adicionar otros hipolipemiantes como ezetimibe, lomitapide, mipomersén e inhibidores de la PCSK9. En los casos en los que no se logren niveles de cLDL aceptables con hipolipemiantes, se requerirán otras terapias como la aféresis.
Estudio genético para el diagnóstico de la HF y el concepto del “caso índice” y del tamizaje “en cascada”Se considera caso índice a la persona a quien inicialmente se le diagnóstica la HF. El tamizaje en cascada se refiere al diagnóstico que se realiza a los familiares en primer grado de consanguinidad del caso índice. Aunque hay controversia sobre si se debe realizar o no el estudio genético en el caso índice y en los familiares (cascada) como requisito primordial para el tratamiento, la recomendación formal de este documento de revisión es que tanto el diagnóstico como la decisión terapéutica sean eminentemente clínicos, y que las pruebas genéticas se hagan en el caso índice para conocer el perfil mutacional, idealmente dentro de un programa nacional de tamizaje de HF y en los casos en los que exista duda diagnóstica. Es preferible realizar el estudio genético cuando el diagnóstico de HF es probable o posible, según los criterios holandeses19. Independiente del estudio genético y ante la sospecha clínica de HF, se debe iniciar tratamiento hipolipemiante en busca del cLDL más bajo posible, para reducir costos e incertidumbre en el paciente y la familia. Algunos argumentos a favor de realizar el estudio genético son: a) Ante la sospecha clínica, el diagnóstico genético es el de certeza, b) Al conocer la mutación en el caso índice se reducen los costos del estudio en cascada, c) Distintos estudios han demostrado que utilizar los niveles de cLDL puede infraestimar o sobreestimar el diagnóstico comparado con el estudio genético, d) Al conocer la mutación se induce mejor adherencia farmacológica y e) El estudio genético es una oportunidad para conocer la prevalencia y las mutaciones en una población2,189,190.
Red iberoamericana de hipercolesterolemia familiarComo se ha mostrado en este documento de revisión, la HF es un trastorno genético relativamente frecuente, que se expresa desde el nacimiento, se manifiesta con hipercolesterolemia importante, enfermedad coronaria prematura y alta morbimortalidad en los sujetos no tratados. Es una entidad subdiagnosticada, subtratada, con buen pronóstico si se detecta y trata de manera precoz y con un arsenal terapéutico en rápida evolución. Por todo ello, la HF constituye un importante problema de salud pública y un reto que debe afrontar el médico.
En consecuencia, en Montevideo, Uruguay, se constituyó la Red Iberoamericana de hipercolesterolemia familiar (RIAHF) el 22 de agosto de 2013, con representantes de Argentina, Brasil, Chile, España, México, Portugal y Uruguay. En septiembre de 2015 fue aceptada Colombia como octavo integrante y se conformó el Capítulo Colombiano de la RIAHF.
Es una organización de carácter multidisciplinario, con participación de médicos y especialidades con experiencia en HF, pertenecientes a países de Iberoamérica. Sus principales objetivos son promover del conocimiento de la HF ante los médicos en general y los servicios de salud, desarrollar mecanismos que permitan un diagnóstico y tratamiento precoces, sensibilizar a los portadores de HF y a su familia, al igual que a las autoridades de salud sobre este problema, impulsar la investigación genética y general para conocer la prevalencia y tipo de mutación específico de manera regional y en Iberoamérica, buscar sinergias y convenios de colaboración, incentivar la educación continuada, y promover criterios diagnósticos homogéneos y registros comunes.
Colombia participará en un registro internacional sobre HF utilizando las bases de datos y los criterios ya establecidos.
Conflictos de interésEste documento fue elaborado con un Grant de la industria farmacéutica destinado a la Educación Continuada, a través de la Sociedad Colombiana de Cardiología y Cirugía Cardiovascular. Los autores declaran haber recibido honorarios para el desarrollo de este documento, sin que por ello, las compañías hayan impuesto alguna condición o solicitaran acceso al mismo antes de su publicación. Igualmente, expresan que no tienen ninguna relación contractual con la industria farmacéutica, en especial con las moléculas que se utilizan en hipercolesterolemia familiar o son nombradas en este documento.
Los coordinadores de este documento, Doctores Alonso Merchán y Álvaro J. Ruiz, expresan sus agradecimientos al Doctor Rodrigo Alonso, profesional experto en hipercolesterolemia familiar, por la asesoría y revisión de este documento.
Documento de revisión sobre hipercolesterolemia familiar (HF), elaborado por la Sociedad Colombiana de Cardiología y Cirugía Cardiovascular, con el apoyo académico de especialistas en Cardiología, Medicina interna, Pediatría, Endocrinología y Genética.