







Las mutaciones en el dominio BCR-ABL1, tirosina quinasa (TK) son mecanismos importantes de resistencia de los inhibidores de la tirosina quinasa (ITK) en pacientes con leucemia mieloide crónica (LMC).
ObjetivoDeterminar el tipo y la frecuencia de las mutaciones en el dominio tirosina quinasa del gen BCR-ABL1, asociadas con falla en la respuesta al tratamiento con imatinib en pacientes con LMC y correlacionar el perfil de mutaciones con los hallazgos clínicos, demográficos, respuesta citogenética y respuesta molecular.
Materiales y métodosSe realizó un estudio descriptivo de tipo prospectivo en pacientes con LMC en tratamiento con IMATINIB a quienes se les realizó cariotipo y análisis de mutaciones del dominio BCR-ABL1 mediante la técnica de PCR anidada.
ResultadosDe los 23 pacientes estudiados en cuatro se encontraron mutaciones: dos presentaron la mutación E255K, uno presentó la mutación H396P y otro presentó doble mutación L387L y T389P. Las mutaciones E255K que se ubican en la región P-loop y H396P en A-loop se asocian con mal pronóstico. La mutación T389P localizada en la región A-loop no está informada en algunas bases de datos.
ConclusionesEn este estudio encontramos cuatro mutaciones en el dominio tirosina quinasa (E255K, H396P, L387L y T389P) que podrían aportar información valiosa y guiar las decisiones de tratamiento. Es importante destacar que esta investigación de análisis mutacional del dominio BCR-ABL es la primera que se realiza en el país con la particularidad adicional de cubrir una población triétnica.
Mutations in the BCR-ABL1 tyrosine kinase domain mutations, are one of the principal mechanisms associated with tyrosine kinase inhibitors (TKI) resistance in patients with chronic myeloid leukaemia (CML).
ObjectivesTo determine the type and frequency of mutations in the tyrosine kinase domain of the BCR-ABL1 gene associated with failure to respond to treatment with Imatinib and Imatinib in patients with CMK, and to correlate the mutation profile with the clinical and demographic variables, as well as the cytogenetic and molecular response.
Materials and methodsA descriptive prospective study was carried out on patients with CML treated with Imatinib. Karyotyping and analysis of the BCR-ABL1 domain mutations were performed on the patients using nested PCR.
ResultsFour types of mutations were found in the 23 patients studied, of which two of them were the E225 mutation, one with the H396P mutation, another with a double mutation L387L and T389P. Both the E255K mutation located in the P-loop region, and H396P mutation in the A-loop region, are associated with a poor prognosis. The T389P mutation located within A-loop region has not been reported in any of the databases.
ConclusionsFour mutations were found in the tyrosine kinase domain (E255K, H396P, L387L and T389P) were found in this study. These findings provide valuable information and as a guideline to help make treatment decisions. It is important to point out that this analytical study on mutations of the BCR-ABL domain is the first one carried out in the country and, specifically, in a tri-ethnic population.
La leucemia mieloide crónica (LMC) es un trastorno mieloproliferativo debido a una expansión incontrolada de las células hematopoyéticas pluripotentes que se caracteriza por la translocación recíproca entre los cromosomas 9 y 22, t(9;22)(q34; q11) conocida como cromosoma Filadelfia (Ph)1, la cual origina el oncogén quimérico BCR-ABL12 que codifica una oncoproteína activa constitutivamente (p210 BCR-ABL tirosina quinasa)3–5 comprometida en la patogénesis de la enfermedad y en su desregulación, condición necesaria y suficiente para la transformación del fenotipo de células LMC6,7. La LMC presenta una incidencia anual en el mundo de 1- 2 casos por cada 100.000 individuos y puede presentarse a cualquier edad, la media de edad de diagnóstico se presenta entre la 5ª y 6ª década de la vida8.
La LMC fue una enfermedad fatal hasta la aprobación clínica en 2001 del imatinib (MI), también conocido como STI571 (Gleevec ®), el cual cambió drásticamente el pronóstico de la enfermedad. Este fue el primer medicamento blanco específico dirigido a la proteína tirosina quinasa BCR-ABL, convirtiéndose rápidamente en el tratamiento estándar de oro en pacientes con LMC que se encontraban en diferentes fases de la enfermedad, cambiando el paradigma en el manejo clínico y la sobrevida9.
El mesilato de imatinib (MI) es un derivado del 2 fenilaminopirimidina que actúa en el dominio tirosina quinasa ocupando en el sitio de unión de nucleótidos en la proteína BCR-ABL y bloquea el acceso al ATP impidiendo la autofosforilación de la tirosina y a su vez, la fosforilación de sus sustratos con la consecuente inactivación de las vías de traducción y señalización10.
El MI se aprobó inicialmente para el tratamiento de primera línea en pacientes con LMC recién diagnosticada en fase crónica (PC) con base en los resultados de eficacia y seguridad del ensayo IRIS (International Randomized Study of Interferón STI571). Sin embargo, un análisis del tipo intención de tratar (intention-to-treat analysis) sugirió que aproximadamente un tercio de los pacientes tratados con imatinib con LMC-CP recién diagnosticada tienen respuestas inadecuadas o no reciben beneficios a largo plazo11–14. Con los primeros casos resistentes a imatinib quedó claro que las células Ph+ podrían evolucionar para eludir la inhibición, llevando a los investigadores a intensos esfuerzos en la caracterización de diferentes mecanismos de resistencia y posteriormente en el desarrollo de nuevos inhibidores. En 2006 y 2007, se aprobaron ITK de segunda generación (nilotinib, NI y dasatinib, DA) inicialmente solo para pacientes con intolerancia a la CML o resistentes al IM. Recientemente, debido a su eficacia para obtener una respuesta citogenética y molecular más rápida, el nilotinib y dasatinib se han utilizado también para la LMC recién diagnosticada15,16. Bosutinib (BO) es el ITK de segunda generación más reciente aprobado para pacientes con CML resistente o intolerante a terapia previa, mientras que ponatinib (PO), un ITK de tercera generación, actualmente solo se puede utilizar en pacientes con mutación T315I o para aquellos con efectos secundarios que surgen durante el tratamiento17.
Con respecto a la resistencia al imatinib se han propuestos varios mecanismos: la evolución cromosómica clonal, la amplificación genómica BCR-ABL1 y las mutaciones en el dominio kinasa (DK)18. Estos mecanismos se presentan en la resistencia primaria o secundaria. La resistencia primaria (intrínseca) se define como la pérdida inicial de respuesta y la resistencia secundaria (adquirida) o recaída como la inestabilidad de la respuesta durante el tratamiento con ITK19. La resistencia en este trabajo se definió con base en los criterios establecidos por European LeukemiaNet (ELN) y las guías de National Comprehensive Cancer Network (NCCN)20.
El término “resistencia a un medicamento” podría ser utilizado cuando este es incapaz de alcanzar el blanco farmacológico, debido a su incapacidad para alcanzarlo (como consecuencia de la disminución biodisponibilidad, inactivación in vivo, interacción negativa con otras sustancias) o alteraciones del objetivo. En la actualidad, la definición aceptada de resistencia a ITKs es el fracaso del tratamiento (resistencia primaria) o cuando se pierde la respuesta lograda (resistencia adquirida o secundaria). La resistencia primaria se puede subdividir en resistencia hematológica primaria que ocurre en 2-4% de los pacientes y resistencia citogenética que ocurre en 15-25% de los pacientes. La resistencia a los ITK en función de los resultados clínicos puede explicarse por mecanismos genómicos, pero también por mecanismos independientes de las mutaciones en BCR-ABL1.
Hasta la fecha, alrededor de 100 diferentes mutaciones del dominio BCR-ABL1 quinasa se han relacionado con IM y, en menor medida, con la resistencia a ITK de segunda generación21–24. Las mutaciones más frecuentes detectadas en pacientes resistentes a IM fueron M244V, G250E, Y253F/H, E255K/V, T315I, F317L, M351T, E355T, F359V y H396R/P25. Entre los pacientes que recayeron en el NI, las mutaciones más frecuentes observadas fueron Y253H, E255K/V, F359V/C /I o T315I, mientras que entre aquellos que recayeron en DA fueron las mutaciones V299L, F317L/V/I/C y T315A/I26.
Hasta la fecha, cerca de 100 diferentes mutaciones del dominio BCR–ABL quinasa se han relacionado con MI y, en menor orden, con la resistencia a ITK de segunda generación y con una prevalencia de 30%-60% en pacientes resistentes a imatinib (fig. 1)27–30. El análisis de mutaciones se indica a los pacientes con resistencia al tratamiento con ITK y estos hallazgos pueden servir de guía para la selección a tratamiento ITKs31. Algunas de esas mutaciones no solamente son resistentes a los ITK de primera generación como imatinib, sino que también son resistentes a los ITK de segunda generación como nilotinib (Y253H, E255/V, F3559V/C/I)32,33 o dasatinib (V299L, T315A, F317L/I/V/C) o ambas34.
 10-20.")
Por otra parte, los pacientes tratados con ITK que presentan mutaciones tienen mayor riesgo de desarrollar mutaciones adicionales, las cuales se asocian con mal pronóstico35,36. El sitio de mutación en DK se localiza principalmente en la región que comprende nueve aminoácidos en posiciones T315I, Y253H/F, MT315T, G250E, E255K/V, F359V y H396R; las cuales presentan diferencias en la sensibilidad a imatinib.
Para el estudio de mutaciones en el DK se han utilizado diversas metodologías tales como: el secuenciamiento directo de Sanger37. Khorashad et al.38, utilizó el pirosecuenciamiento para el estudio de 305 pacientes con LMC, Mascarenhas CC et al.39, analizaron 93 pacientes mediante cromatografía líquida de alta resolución HPLC, Machova K et al.40 emplearon la técnica High-resolution melt curve analysis para estudiar 101 pacientes con LMC e identificar mutaciones en el dominio tirosina quinasa. Por otra parte, la reciente aparición de las tecnologías de nueva generación (NGS) como las expuestas por Soverini et al.41 ofrecen oportunidades sin precedentes para orientar nuevos estudios en leucemias debido a: 1) Su alta sensibilidad de la detección de mutaciones, manteniendo al mismo tiempo la posibilidad de caracterizar completamente el espectro de las mutaciones de variantes menores. 2) Permite reconstruir la arquitectura clonal mutante cuando hay múltiples mutaciones, lo que facilita distinguir las diferentes poblaciones. 3) Permite cuantificar la carga de la población mutante y seguir la dinámica en el tiempo. El objetivo de este estudio fue identificar el punto de mutación en dominio tirosina quinasa ABL en pacientes con falla o pérdida de la respuesta a imatinib y determinar la asociación con características clínicas y la influencia de las mutaciones en el desarrollo de la enfermedad.
El objetivo de este estudio es determinar el sitio, tipo y la frecuencia de mutaciones en el dominio tirosina quinasa del gen BCR-ABL1 asociado a la falta de respuesta al tratamiento con imatinib e imatinib seguidos por inhibidores de segunda generación después de la resistencia en pacientes con CML y correlacionar el perfil de mutación con los hallazgos clínicos, demográficos, respuesta citogenética y respuesta molecular.
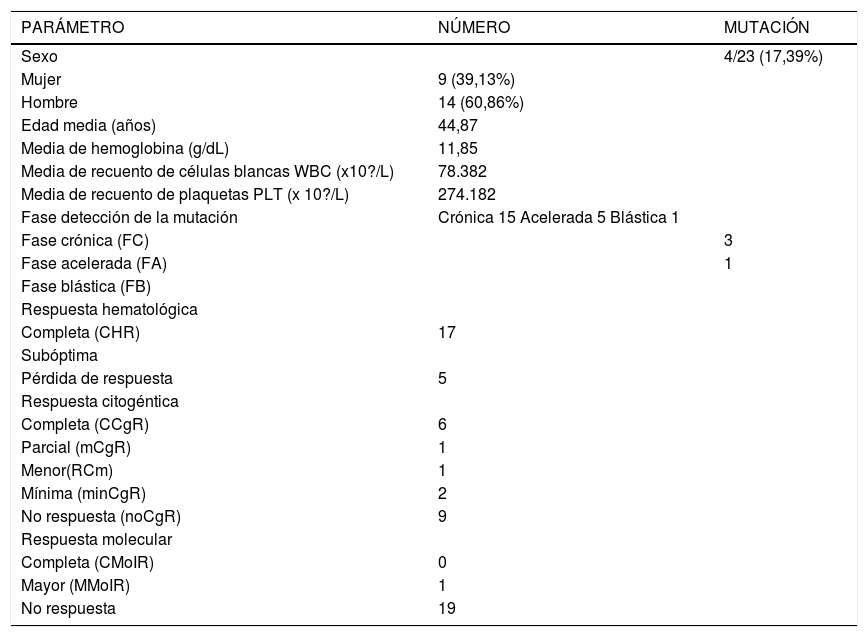
Pacientes, muestras y métodosSe realizó un estudio descriptivo de tipo prospectivo, las muestras se obtuvieron a conveniencia y con previo diagnóstico clínico, hematológico, citogenético de LMC y con terapia de imatinib. Además, debían cumplir los criterios de inclusión de resistencia descritos por Bacarani, remisión hematológica completa WBC< 10 X109 /L, remisión citogenética complete (RCgC) ausencia de filadelfia Ph+, respuesta parcial (RPCg), metafases del 1% a 35% Ph+, respuesta menor (RCgm) metafases del 36% al 65% Ph+, respuesta mínima, metafases mínimas (RCgmin) del 66% al 95% y no respuesta citogenética (RCgn) >95% de metafase Ph+. La población de estudio fue constituida por 23 pacientes, 14 hombres y 9 mujeres. Las muestras de médula ósea o de sangre periférica de los pacientes fueron obtenidas por los hematólogos en diferentes instituciones de salud y remitidas al laboratorio de Genética Médica de la Facultad de Medicina de la Universidad de Antioquia, durante 2010 y 2012. Los pacientes firmaron el consentimiento aprobado por el Comité de Bioética, Sede de Investigación Universitaria de la Universidad de Antioquia, de la ciudad de Medellín. Las características clínicas y demográficas, respuesta citogenética, respuesta molecular y estado mutacional de los pacientes se indican en la tabla 1.
Características clínicas y demográficas, respuesta citogenética, respuesta molecular y estado mutacional de los pacientes con LMC
| PARÁMETRO | NÚMERO | MUTACIÓN |
|---|---|---|
| Sexo | 4/23 (17,39%) | |
| Mujer | 9 (39,13%) | |
| Hombre | 14 (60,86%) | |
| Edad media (años) | 44,87 | |
| Media de hemoglobina (g/dL) | 11,85 | |
| Media de recuento de células blancas WBC (x10?/L) | 78.382 | |
| Media de recuento de plaquetas PLT (x 10?/L) | 274.182 | |
| Fase detección de la mutación | Crónica 15 Acelerada 5 Blástica 1 | |
| Fase crónica (FC) | 3 | |
| Fase acelerada (FA) | 1 | |
| Fase blástica (FB) | ||
| Respuesta hematológica | ||
| Completa (CHR) | 17 | |
| Subóptima | ||
| Pérdida de respuesta | 5 | |
| Respuesta citogéntica | ||
| Completa (CCgR) | 6 | |
| Parcial (mCgR) | 1 | |
| Menor(RCm) | 1 | |
| Mínima (minCgR) | 2 | |
| No respuesta (noCgR) | 9 | |
| Respuesta molecular | ||
| Completa (CMoIR) | 0 | |
| Mayor (MMoIR) | 1 | |
| No respuesta | 19 |
Nomenclatura: CCgR: no metafases Ph+. mCgR: de 1% a 35% de metapases Ph+. RCm: de 36% a 65% de metafases Ph+. minCgR: de 66% a 95% de metafases Ph+. noCgR: más de 95% de metafases Ph+. No respuesta: metafases Ph+ >95%.
CMoIR: no se detecta transcrito BCR-ABL en las pruebas de PCR. MMoIR: relación BCR-ABL y ABL >/= 0,1%. Pérdida de la respuesta molecular BCR-ABL > 0,1%(IS)
El análisis cromosómico se realizó mediante la técnica estándar de bandas G y las alteraciones cromosómicas se describieron con base en el Sistema Internacional de Nomenclatura en Citogenética- ISCN 201342. Para la extracción del ARN total se utilizó RNeasy® Mini Kit QIAGEN, la síntesis de y la detección de cuantificación de BCR-ABL t(9;22) con el kit Roche Diagnostics LightCycler, siguiendo las instrucciones del fabricante. Este proceso se llevó a cabo en un termociclador LightCycler® 2.0 Roche y el diagnóstico de la fusión génica BCR-ABL se realizó mediante la amplificación del dominio tirosina quinasa por PCR anidada.
Amplificación de dominio kinasa BCR-ABL por PCR semianidadaLos productos de la PCR se prepararon utilizando un par de primers diseñados para cubrir gen de fusión BCR-ABL1 (Exón 2 BCR al exón 10 ABL1) con un producto esperado de 1.600 pb. Una segunda ronda de amplificación PCR anidada para el dominio TK (ABL1 exón 4 al 10), con un producto de 863pb (47). El ADNc obtenido se utilizó como molde en la primera PCR con los siguientes par de primers Forward BCR/Ex13-F 5’ TTCAGAAGCTTCTCCCTGACAT 3’ y Reverse Primer ABL1/Ex10-5’GTACTCACAGCCCCACGGA 3’. El volumen final de la reacción fue de 25μL compuesto por: 0,2 U de ADN Taq polimerasa (Thermo Scientic reconvinanate), buffer 5X 2,5mM MgCl2, 200μM dNTPs, 0,5μM de cada primer y 2μL ADNc. La reacción se llevó a cabo en un termoclicador (Veriti, Applied Biosystems, CA).
Las condiciones de la primera PCR fueron las siguientes: 1 ciclo inicial a 95°C durante 3min, seguido de 35 ciclos a 95°C durante 30 segundos, 60°C x 40 s, y 72°C durante 90 s. La reacción se finalizó con un paso adicional de extensión a 72°C de 5min. El producto amplificado de esta reacción sería de 1.600 (pb). El producto de amplificación de la primera PCR se diluyó 1:1000 y se empleó como molde de la segunda reacción de amplificación. En esta segunda reacción se utilizó el primer Forward primer ABL1/Ex4-F 5’ AAGCGCAACAAGCCCACTGTCTAT 3’ y el mismo primer antisentido de la primera reacción. El perfil térmico de la PCR empleado fue el siguiente: 1 ciclo a 95°C durante 3min, seguido de 35 ciclos a 95°C durante 30 s, 60°C x 40 s y 72°C durante 60 s, y se finalizó también con un paso adicional de extensión de 5min. El producto de PCR 865 pb se visualizó en geles de agarosa al 1,5% en tampón TAE 1X (40mM de tris-acetato, 1mM EDTA (pH 8,0)) de manera similar, a como se mencionó anteriormente. En ambos procesos de amplificación se utilizó el kit de Taq DNA Polymerase recombinant 5 U/μL (Thermo Scientific), como control positivo se tomó ADNc obtenido del cultivo celular K562 y como control negativo el ADNc de un voluntario sano.
Secuenciamiento de las muestrasLos productos de PCR obtenidos en la segunda reacción se enviaron a secuenciar por ambas cadenas a MACROGEN INC (Seúl, Corea del Sur), con los mismos primers empleados en la respectiva reacción. Los resultados del secuenciamiento se visualizaron y se editaron empleando el software Chromas Lite y la posición de los nucleótidos se realizó mediante comparación con la secuencia consensus del cDNA GenBank NM - 005157 y para la determinación de mutaciones se utilizó la base de datos COSMIC (Catalog of Somatic Mutations in Cancer)43. El análisis de bioinformática se realizó con el programa PolyPhen-2 para predecir las propiedades funcionales de la nueva mutación T389P con base en la secuencia de referencia NP - 005148.2
Análisis estadísticoLos resultados se analizaron mediante el programa SPSS versión 19. Se realizó un estudio descriptivo prospectivo de una sola medición por paciente. En la estadística descriptiva para las variables cuantitativas se utilizó la media y el rango; en las variables categóricas se presentaron frecuencias y porcentajes. Los datos para la edad y parámetros hematológicos de pacientes tratados con imatinib e imatinib más otros inhibidores de tirosina quinasa de segunda generación se determinaron mediante la prueba de Shapiro-Wilk y por la prueba de Levene, los cuales indicaron una distribución normal y homocedasticidad entre los diferentes grupos. Debido a que tanto el supuesto de normalidad como el de homocedasticidad se cumplieron, las comparaciones de las medias se realizaron mediante la prueba t de Student para muestras independientes.
ResultadosSe realizó el análisis de mutaciones en 23 pacientes con LMC tratados con inhibidores de tirosina quinasa, 15 hombres y 8 mujeres. La edad media de los pacientes tratados con imatinib y los tratados con inhibidores de segunda generación fue 48 y 43 años (p=0,191) respectivamente, mientras que la media entre los pacientes con mutaciones o sin estas fue de 54 y 42 años respectivamente (p=0,423). La mayoría de los pacientes estaban en fase crónica, solo uno con mutaciones estaba en fase acelerada. El valor de la media del recuento de glóbulos blancos entre los pacientes tratados con imatinib y los tratados con los ITKs de segunda generación fue significativo (p=0,001). Con respecto a los valores de hemoglobina y plaquetas no presentaron diferencias significativas. Por otra parte, al comparar los parámetros anteriores entre el grupo de pacientes con mutaciones y no mutados se hallaron valores estadísticamente significativos para glóbulos blancos y plaquetas (p=0,000) y (p=0,003) respectivamente.
Con respecto a la terapia todos los 23 pacientes fueron tratados al inicio con imatinib 400mg/*. Ocho de estos pacientes solo recibieron GLEEVEC® (Imatinib mesylate) durante todo el tiempo. De los 15 pacientes restantes que presentaron falla al tratamiento, se cambiaron a inhibidores de segunda generación (tabla 2). Con respecto a la fase de diagnóstico, 15 pacientes se encontraban en fase crónica, 5 en fase acelerada y 1 en fase blástica. La respuesta hematológica completa (RHC) la alcanzaron 6 pacientes y 14 presentaron pérdida de la respuesta hematológica completa (RHC), 1 presentó respuesta citogenética parcial. Sin respuesta molecular (RMC) 19 pacientes, solo 1 con RMC.
Frecuencia de mutaciones ABL en el dominio TK en pacientes con LMC
| Tipo de paciente con LMC | Número de casos estudiados | Número de casos con mutaciones en el dominio TK (%) | Tipos de mutación |
|---|---|---|---|
| Expuestos solo a imatinib | 8 | 1 | H396P A - Loop |
| Expuestos a imatinib y a ITK de segunda generación | 15 | 3 | T389P L187L E255K P - Loop |
De los 23 pacientes estudiados solo en cuatro se encontraron mutaciones (17,4%), dos presentaron la mutación E255K (8,6%) (fig. 2), uno presentó la mutación H396P (4,3%) (fig. 3), y uno presentó doble mutación L387L y T389P (4,3%) (fig. 4). Las mutaciones E255K localizada en la región P-loop y H396P en A-loop se han informado en pacientes con falla al tratamiento con ITK debido a su insensibilidad al imatinib y sensibilidad intermedia a dasatinib y nilotinib respectivamente; ambas mutaciones se asocian con mal pronóstico44.
.")

.")
Con respecto a la mutación L387L localizada en la región A-loop es una mutación sinónima o silenciosa y no impide la unión del imatinib. Al contrario, la mutación T389P localizada en la región A-loop, induce a un cambio en la posición 389 de (A>C) Treonina (T) por Prolina (P). Esta sustitución no se ha informado en las bases de datos Cosmic, Esembl y NCBI; solo se encuentra como un SNP T389T que corresponde a la referencia rs752694546 en la base de datos de NCBI. Esta mutación podría considerarse como nueva en pacientes con LMC resistentes a imatinib. En la figura 1, se muestra la nueva mutación T389P DTK. Cabe anotar, que uno de los pacientes con mutación E255K estaba en fase acelerada, con respuesta hematológica completa y resistente a imatinib, los demás estaban en fase crónica.
DiscusiónEste es el primer estudio en Colombia sobre la detección de mutaciones en DTK en pacientes con LMC tratados con imatinib que tiene como fin contribuir al conocimiento de la patogénesis y del comportamiento de esta enfermedad en nuestro medio.
El mecanismo de resistencia más frecuente en pacientes con LMC tratados con imatinib es la mutación puntual del dominio ABL quinasa que afecta directa o indirectamente la unión de los ITK al bolsillo de la quinasa alterando la conformación del BCR-ABL disminuyendo afinidad al inhibidor específico o interfiriendo con el sitio de unión a ITK. Las mutaciones se localizan en diferentes subunidades estructurales del dominio quinasa y se clasificaron en cuatro categorías: (1) el sitio de unión directa, (2) el dominio de unión a fosfato (bucle P), (3) el dominio de activación (bucle A), y (4) el dominio catalítico (C-loop). La adquisición de mutaciones puntuales en el dominio de tirosina quinasa ABL de BCR-ABL se observa en más del 50% de pacientes con LMC con resistencia clínica y se encuentra con mayor frecuencia en pacientes con resistencia adquirida que resistencia primaria45–48.
Asimismo, el desarrollo de la resistencia a imatinib (IM) en pacientes con LMC se relaciona con mecanismos heterogéneos específicos que incluyen las vías dependientes e independientes del BCR-ABL1. La vía dependiente se ha informado con más frecuencia en los pacientes con LMC resistentes a IM y comprende principalmente las mutaciones en DTK en el gen BCR-ABL1 Druker et al.49, demostraron que el bloqueo de la proteína BCR-ABL1 en la conformación inactiva por IM previene la transferencia del grupo fosfato del AMP al sustrato ATP e inhibe la vía de señalización corriente abajo. Sin embargo, Corbin et al.50, indicaron que algunas mutaciones pueden alterar el punto crítico de unión entre el IM y el BCR-ABL1, además otras mutaciones parecen inducir una transición de la conformación inactiva a la activa, a la que el IM no se podrá unir
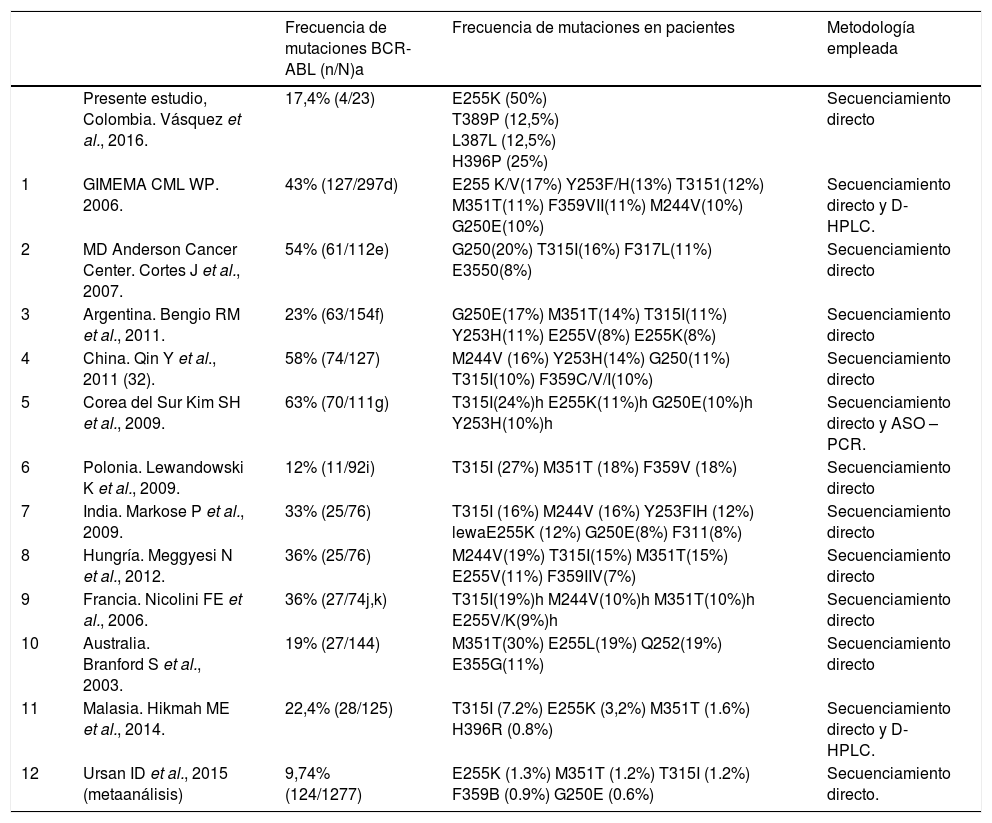
En el mundo y principalmente en países desarrollados se han publicado estudios de mutaciones en DK, BCR-ABL, pero son pocos los que exploran las frecuencias y el tipo de mutación en el DK. En este trabajo se analizaron los resultados con respecto a las frecuencias de mutaciones en 12 investigaciones informadas en la literatura. Cabe anotar que en todas ellas se utilizó imatinib como tratamiento de primera línea y el método análisis utilizado para identificar las mutaciones fue el secuenciamiento directo (Sanger).
Ursan et al. realizaron un metaanálisis sobre características clínicas e inhibidores de ITK asociados a LMC en dominio BCR-ABL51, informaron en su trabajo que las mutaciones más comunes fueron E225K (1,3%), M351T (1,2%), T315I (1,2%), F359B (0,9%) y G250E (0,6%). Al compararlos con los resultados obtenidos en nuestro trabajo, podemos apreciar que la mutación más común en nuestro trabajo correspondió a la mutación E255K (50%), comparada con el estudio anterior (1,3%) la nuestra fue muy superior. Por otra parte, ninguna de las demás mutaciones informadas por Ursan et al. fueron halladas en nuestro trabajo. Además, las mutaciones encontradas en los 12 trabajos analizados, incluyendo el anterior se informan en la tabla 3, donde puede apreciar que la mutación más frecuente corresponde a la E255K, en un rango entre 1,3% a 50%. Este valor del 50% se encontró en nuestro trabajo seguido por GIMEMA (17%) Markose P et al. (12%), Kin SH (11%), Nicoline et al. (9%), Hikmah ME (3,2%) y Ursan et al. (1,3%), respectivamente. La segunda mutación más frecuente fue M351T presente en un rango entre 1,2% al 18%. La menor frecuencia se informó en el trabajo publicado por Ursan et al. (1,2%), siguiendo en orden ascendente por Hi Kmah ME (1,6%), Nicoline et al. (10%), GIMEMA (11%), Bengio et al. (14%), Meggyesi et al. (15%) y finalmente Lewanadowski et al. (18%). Esta mutación no se encontró en nuestro trabajo.
Presentación de frecuencias de mutaciones en DK de BCR-ABL en pacientes resistentes expuestos a inhibidores de tirosina quinasa informados en diferentes poblaciones. Tabla adaptada de Soverini et al. 2013(63)
| Frecuencia de mutaciones BCR-ABL (n/N)a | Frecuencia de mutaciones en pacientes | Metodología empleada | ||
|---|---|---|---|---|
| Presente estudio, Colombia. Vásquez et al., 2016. | 17,4% (4/23) | E255K (50%) T389P (12,5%) L387L (12,5%) H396P (25%) | Secuenciamiento directo | |
| 1 | GIMEMA CML WP. 2006. | 43% (127/297d) | E255 K/V(17%) Y253F/H(13%) T3151(12%) M351T(11%) F359VII(11%) M244V(10%) G250E(10%) | Secuenciamiento directo y D-HPLC. |
| 2 | MD Anderson Cancer Center. Cortes J et al., 2007. | 54% (61/112e) | G250(20%) T315I(16%) F317L(11%) E3550(8%) | Secuenciamiento directo |
| 3 | Argentina. Bengio RM et al., 2011. | 23% (63/154f) | G250E(17%) M351T(14%) T315I(11%) Y253H(11%) E255V(8%) E255K(8%) | Secuenciamiento directo |
| 4 | China. Qin Y et al., 2011 (32). | 58% (74/127) | M244V (16%) Y253H(14%) G250(11%) T315I(10%) F359C/V/I(10%) | Secuenciamiento directo |
| 5 | Corea del Sur Kim SH et al., 2009. | 63% (70/111g) | T315I(24%)h E255K(11%)h G250E(10%)h Y253H(10%)h | Secuenciamiento directo y ASO – PCR. |
| 6 | Polonia. Lewandowski K et al., 2009. | 12% (11/92i) | T315I (27%) M351T (18%) F359V (18%) | Secuenciamiento directo |
| 7 | India. Markose P et al., 2009. | 33% (25/76) | T315I (16%) M244V (16%) Y253FIH (12%) lewaE255K (12%) G250E(8%) F311(8%) | Secuenciamiento directo |
| 8 | Hungría. Meggyesi N et al., 2012. | 36% (25/76) | M244V(19%) T315I(15%) M351T(15%) E255V(11%) F359IIV(7%) | Secuenciamiento directo |
| 9 | Francia. Nicolini FE et al., 2006. | 36% (27/74j,k) | T315I(19%)h M244V(10%)h M351T(10%)h E255V/K(9%)h | Secuenciamiento directo |
| 10 | Australia. Branford S et al., 2003. | 19% (27/144) | M351T(30%) E255L(19%) Q252(19%) E355G(11%) | Secuenciamiento directo |
| 11 | Malasia. Hikmah ME et al., 2014. | 22,4% (28/125) | T315I (7.2%) E255K (3,2%) M351T (1.6%) H396R (0.8%) | Secuenciamiento directo y D-HPLC. |
| 12 | Ursan ID et al., 2015 (metaanálisis) | 9,74% (124/1277) | E255K (1.3%) M351T (1.2%) T315I (1.2%) F359B (0.9%) G250E (0.6%) | Secuenciamiento directo. |
Con respecto a la mutación T315I se encontró en un rango entre el 1,2% al 27%. La menor frecuencia 1,2% fue informada por Ursan et al. siguiendo de igual forma en orden ascendente por Hikmah ME (7,2%), Quin Y et al. (10%), Bengio et al. (11%), GIMEMA (12%), Meggyesi et al. (15%), Cortes et al. (16%), Markose P et al. (16%), Nicolini FE et al. (19%), Kin SH et al. (24%) y finalmente Lewanadowski K et al. (27%). Por otra parte, cabe resaltar que la mutación H396P se encontró en nuestro trabajo en un 25%, y también la informó Hikmah ME et al. pero con una variación H396R (0,8%). Las mutaciones T389P (12,5%) y L387L (12,5%) encontradas en nuestro trabajo no se informan en las publicaciones anteriores.
Estas diferencias en las frecuencias de mutaciones se pueden deber a varios factores: 1. El número de individuos de estas poblaciones fueron mucho mayor que el nuestro. 2. El número de pacientes encontrados en la fase acelerada fue mayor, en esta fase donde es más probable encontrar mutaciones, lo que podría indicar que las mutaciones en DK se originan por inestabilidad genómica durante el curso natural de la enfermedad, así esto podría soportar el hecho que en nuestro trabajo el 13% de los pacientes con mutaciones se encontraban en fase crónica y uno en fase acelerada; opuesto a lo informado en la literatura, lo que es explicable por el bajo número de pacientes reclutados en este trabajo. 3. Además, las variaciones en las frecuencias de mutaciones en el DK y el tipo de mutación pueden reflejar la heterogeneidad entre las poblaciones étnicas estudiadas. Por otra parte, es importante anotar que en una paciente se encontró una doble mutación, la L387L que corresponde a un cambio en la posición 1161 G>A, la cual se considera una mutación silenciosa y con baja frecuencia; solo ha sido informada por Brandford y Hughes en 201052. Con respecto a la mutación T389P (A>C) no se ha informado en las bases de datos consultadas, por lo tanto, corresponde a una nueva mutación. Cabe anotar que esta paciente a la fecha está viva, pese a presentar esta doble mutación y que en algunas ocasiones no recibe a tiempo los medicamentos.
Otro aspecto importante es la ubicación de las mutaciones en el dominio kinasa, las mutaciones ubicadas en la región P – loop (E255K) se asocian con corta sobrevida, lo que puede explicarse por la presencia de pequeños clones con mutaciones y que se tornan más agresivos con el tiempo. Bajos niveles de clones con mutaciones específicas pueden tener diferente significado clínico. De los dos pacientes con la mutación E255K, uno falleció. Por otra parte, el paciente con resistencia al tratamiento y sin mutaciones, falleció, y la paciente con la mutación H396P aún está viva.
Diferentes mutaciones pueden presentar variación con respecto a la resistencia ITK dependiendo de su localización y el efecto sobre la TK. Las mutaciones encontradas en este estudio pueden ser categorizadas en tres grupos con base en el grado de sensibilidad para el IM, por ejemplo, sensible (H396P, L387L), insensible E255K y la tercera según O’Hare et al.53, con sensibilidad intermedia tanto para dasatinib y nilotinib, o según Redaelli et al.54, como resistente La mutación H396P es sensible tanto para dasatinib como nilotinib55 y resistente56; y la nueva mutación T389P analizada en el programa Polyphen 2.0 muestra un score 0,832, sensibilidad de 0,84 y especificidad de 0,93 por lo cual se considera de alta penetrancia y mal pronóstico.
En total se identificaron 4 mutaciones y una de ellas fue nueva. Estas mutaciones presentaron varios tipos de sensibilidad tanto a los inhibidores de primera y segunda generación, las cuales se han informado en estudios de otras poblaciones. Así mismo, deberían realizarse grandes investigaciones clínicas con los diferentes inhibidores de TK con el fin de conocer el impacto real de estas mutaciones en nuestra población. Además, se debe prestar atención cuando no se detectan mutaciones en DK para explicar la resistencia a ITKs por fuera del DK en regiones cercanas, cuando hay evolución cromosómica clonal, amplificación BCR-ABL y variaciones farmacogenómicas. La información reciente sugiere que las células madre de LMC son capaces de sobrevivir a pesar de la inhibición de BCR-ABL, lo que indica que la inhibición de los clones leucémicos puede necesitar estrategias completamente diferentes.
En general, los pacientes portadores de clones resistentes a TKI parecen estar en mayor riesgo de acumular aberraciones genómicas adicionales que conducen a la progresión a crisis blástica. Particularmente, los pacientes resistentes a MI tenían una probabilidad más alta de retraso asociado con el desarrollo de más mutaciones comparados con los pacientes que no la tenían.
Igualmente, en los pacientes con células BCR-ABL1 transformadas exhiben proliferación celular desregulada, independencia de los factores de crecimiento y apoptosis como resultado de una intrincada red de señalización, vías de acceso y la represión estricta de la vía homeostática molecular que controla el crecimiento y diferenciación de los progenitores hematopoyéticos. Sin embargo, está claro que la quinasa BCR-ABL1 actúa independiente de los factores de crecimiento y la apoptosis; pero interviene en múltiples eventos celulares y genéticos que se acumulan con el tiempo para inducir la enfermedad a la fase blástica. El estudio de mutaciones en estos pacientes podría ampliar el entendimiento de los mecanismos de resistencia y aumentar nuestra capacidad para mejorar el tratamiento con los ITKs en pacientes con mutaciones resistentes.
Con nuestros hallazgos, recomendamos profundizar en el campo de la genética y biología molecular de las mutaciones en pacientes con leucemia mielode crónica, con el fin de aumentar la comprensión de los mecanismos que regulan la progresión a la fase acelerada, la resistencia a ITK, la quiescencia de las células madre, la inactivación de los genes supresores de tumores, así como la inestabilidad genómica y reparación del ADN, son esenciales para desarrollar estrategias de tratamiento para los pacientes con LMC.
En la actualidad se realiza la integración del análisis de mutaciones en el algoritmo de tratamiento para los pacientes con LMC, esto hace parte de las nuevas guías de tratamiento (CNNC 2017) (57), en las cuales se recomiendan el estudio citogenético y cuantificación molecular p210 al diagnóstico y seguimiento. En el caso de resistencia se solicita el análisis de mutaciones, lo que permitiría una mejor selección de la estrategia de tratamiento de segunda línea, para la cual se deben tener en cuenta otros factores como la historia del paciente, factores de riesgo, comorbilidad que conlleva a una mayor supervivencia del paciente y disminución de los costos de tratamiento.
Este trabajo aporta información relevante que se puede utilizar como un componente importante del proceso de toma decisiones y proporciona orientación adicional a los clínicos sobre la mejor manera de integrar la identificación de las diferentes mutaciones, la fase de la enfermedad en que se presenta, de la baja respuesta o falla al tratamiento de cada paciente, con el fin de someterlos a inhibidores de segunda o tercera generación como ponatinib o tomar la decisión de realizarle trasplante de médula ósea. Por último, es importante resaltar que nuestro estudio sería el primero en el país y específicamente en una población étnica trirracial.
Fuentes de financiaciónEste trabajo de investigación fue financiado por la Universidad de Antioquia, Medellín, Colombia. Programa de Sostenibilidad de Grupos 2009-2010, Genética Médica, CPT-0318 y Novartis SA. Colombia.
Conflicto de interesesLos autores declaran que no tienen conflicto de intereses.
Los autores agradecen la colaboración de la Dra. Susan Branford por corroborar las mutaciones en su laboratorio, a la Dra. Beatriz Aristizábal del Hospital Pablo Tobón Uribe donde se realizó la cuantificación del trascripto P210 y hemograma de los pacientes. A la Unidad de Genética Médica, Facultad de Medicina Universidad de Antioquia, donde se realizaron los estudios citogenéticos. Al Dr. Mauricio Camargo y la bióloga Olga Lucía por colaborar con algunos pacientes para el estudio, a los Doctores José domingo Saavedra y Fabiola Bizcarra de Clínica Vida y Amado Kardus de Clínica Las Américas de Medellín. Al Dr. Jorge Botero Garcés por su permanente asesoría en los análisis estadísticos y a la ingeniera biológica Katherine Palacio y Dr. Nicolás Pineda por su colaboración en el análisis de secuencias e identificación de mutaciones.
Este trabajo de investigación fue financiado por la Universidad de Antioquia, Medellín, Colombia. Programa de Sostenibilidad de Grupos 2009-2010, Genética Médica, CPT-0318 y Novartis SA. Colombia.
