






El síndrome reproductivo y respiratorio porcino (PRRS) es una enfermedad viral endémica en las regiones porcinas del mundo, y su control es deficiente. La rápida identificación de las especies PRRSV-1 y PRRSV-2 es de gran importancia para la vigilancia epidemiológica molecular del virus. El objetivo de este estudio fue la caracterización molecular del gen ORF5, que codifica la proteína glicosilada 5 (GP5), del agente causal del virus causante del PRRS detectado en granjas porcinas de Lima, Perú. De 100 muestras de suero porcino, 24 fueron positivas para el PRRSV-2, lo que fue confirmado mediante 2 técnicas moleculares. La inferencia filogenética demostró la relación parental con la variante NADC34 y se observó una mejor relación filogenética de las cepas en estudio con aislados peruanos de referencia en el linaje 1 y sublinaje 1.5 y con las cepas patógenas de diversas partes del mundo. Las cepas 25 a 35, 40, 41 y 43 a 48 tuvieron valores altos de homología nucleotídica (95-98%) y aminoacídica (92-98%), mientras que las cepas 36, 37, 38 y 39 mostraron una alta variabilidad genética, con valores bajos de homología nucleotídica (83%) y aminoacídica (79%), y la cepa 42 tuvo homologías del 89 y 84% en relación con la variante NADC34. En conclusión, demostramos la eficacia en la identificación de los cebadores in house validados en comparación con un kit comercial, además de la evolución y dispersión del linaje 1 sublinaje 1.5 y la relación filogenética con la variante NADC34 de la especie PRRSV-2 en Lima, Perú.
The porcine reproductive and respiratory syndrome (PRRS) is an endemic disease in pork-producing regions of the world, and its control remains poor. Rapid identification of PRRSV-1 and PRRSV-2 species is of great importance for molecular epidemiological surveillance of the virus. The objective of this study was the molecular characterization of the ORF5 gene that synthesizes glycosylated protein 5 (GP5) from PRRS virus detected in pig farms in Lima, Perú. One hundred serum samples were used, obtaining 24 PRRSV-2-positive strains confirmed by two molecular techniques. Phylogenetic inference demonstrated the parental relationship with the NADC34 variant, and we achieved a better phylogenetic relationship of the study strains with Peruvian reference isolates in lineage 1 and sublineage 1.5 and pathogenic strains from various parts of the world. Homology analysis showed that strains 25 to 35, 40 to 41, and 43 to 48 obtained high nucleotide homology values of 95-98% and amino acid homology of 92-98%; strains 36, 37, 38, and 39 with high genetic variability with low nucleotide homology values of 83% and amino acid homology of 79%, and strain 42 with homology of 89 and 84% in relation to the NADC34 variant. In conclusion, we demonstrated the effectiveness of the validated in-house primers for identification compared to a commercial kit, in addition to the evolution and dispersion of lineage 1, sublineage 1.5 and the phylogenetic relationship with the NADC34 variant of the PRRSV-2 species in Lima, Perú.
El virus del síndrome reproductivo y respiratorio porcino (PRRSV) es causante de brotes con alta tasa de mortalidad en todo el mundo, generando pérdidas económicas de millones de dólares en la industria porcina1,5. La enfermedad se caracteriza por abortos, nacimiento de lechones débiles, mortalidad de lactantes y enfermedades respiratorias en cerdos de mediana edad4,22. El virus es parte de la familia Arteriviridae, subfamilia Variarterivirinae, género Betaarterivirus y subgéneros Ampobartevirus y Eurpobartevirus3,6. Se reconocen dos especies, Betaarterivirus americense y Betaarterivirus europensis, con aproximadamente un 60% de similitud entre sí3,15. La partícula viral mide entre 50 y 74nm, es de simetría icosaédrica, su envoltura cubre una nucleocápside esférica interna de 39nm de diámetro y su genoma está representado por una molécula de ARN lineal simple, de aproximadamente 15kb, con polaridad positiva3. El genoma viral contiene al menos 10 marcos de lectura abiertos: ORF1a, ORF1b, ORF2a, ORF2b, ORF3, ORF4, ORF5, ORF5a, ORF6 y ORF724,27.
El gen ORF5 codifica la principal glicoproteína de la envoltura viral, la GP5. Esta proteína se encuentra en la superficie y es necesaria para la unión del virus a los receptores celulares2,24. El ORF5 es uno de los genes más variables del genoma viral y a menudo se utiliza en análisis filogenéticos como marcador molecular para rastrear la mutación del virus14.
En trabajos previos, el análisis filogenético del gen ORF5 en la especie Betaarterivirus americense 2 dio como resultado 9 linajes monofiléticos, L1 a L9, y 3 linajes específicos de Corea (KOR A, KOR B y KOR C)16,17. Esta gran diversidad de variantes debido a la alta heterogeneidad genética y variabilidad expuesta en la glicoproteína 5 respalda la teoría de que las variantes del PRRSV pueden adquirir de forma inherente distintos patrones de virulencia y presentaciones clínicas13, además de jugar un papel importante en la infectividad viral por importantes dominios inmunológicos asociados con la neutralización, como el epítopo neutralizante viral8,9,23.
En el Perú, se desconoce el año de ingreso del PRRSV, pero se sabe que a finales de la década de 1990 se incrementó la frecuencia de problemas respiratorios y reproductivos porcinos en granjas, principalmente en la zona de Lima19. Por esta razón, fue necesario identificar los linajes y sublinajes presentes en nuestro país, para proporcionar una referencia científica para la prevención, la detección y el control mediante vacunas legales por SENASA. El objetivo de esta investigación fue efectuar la caracterización molecular de las especies del PRRSV circulantes en granjas porcinas de Lima, aplicando vigilancia epidemiológica molecular para la identificación del patógeno por un diseño in house de RT-PCR convencional y secuenciación del gen ORF5. También se exploraron las relaciones genéticas con las secuencias del PRRSV originarias de otros países americanos, latinos, europeos y asiáticos disponibles en GenBank. Los resultados de este estudio proporcionan una comprensión de la variación genética del virus y demuestran variantes circulantes en nuestro país por técnicas moleculares.
Materiales y métodosMaterial biológicoLos veterinarios de las granjas realizaron un muestro por conveniencia debido a las limitaciones logísticas y presupuestarias. Ellos determinaron como criterios la accesibilidad y tenencia de cerdos con signos clínicos compatibles con el PRRS en los años 2020 y 2021 en la provincia de Lima. Se recibieron 100 muestras de sangre de un total de 12 granjas; cada representante trajo un promedio de 9 tubos, algunos se eliminaron por no cumplir con los requisitos para el procedimiento. Las muestras se transportaron en cajas de hielo seco hermetizadas al laboratorio IMBios de Montana y se centrifugaron en el equipo Orto Alresa modelo Cónsul 21 a 2.500×g×5min. Los sueros resultantes fueron trasvasados a tubos de 1,5ml y congelados a −20°C.
Extracción y cuantificación del ARNEl ARN viral fue extraído utilizando el InnuPREP virus DNA/RNA kit (Analytik Jena), siguiendo el instructivo del producto. Para esto, se utilizaron 200μl de muestras de suero y vacuna inactivada de tipo 2 (americano) cepa Nebraska Prime Pac® PRRS y vacuna inactivada tipo 1 (europeo) cepa VP-046 SUIPRABAC® PRRS. Se verificó la pureza mediante la cuantificación por espectrofotómetro, siendo la relación A260-A280 de lectura similar a 2 en la mayoría de las muestras. El ARN fue almacenado a −80°C.
Retrotranscripción en tiempo real (RT-qPCR) de IDEXX para la detección de especies de Betaarterivirus americense y Betaarterivirus europensisLas muestras de suero de campo fueron analizadas siguiendo el instructivo del RealPCR* NA PRRS Type 1 and Type 2 Multiplex RNA Test de IDEXX, usado para detectar y diferenciar el tipo 1 (europeo) y el tipo 2 (norteamericano) del ARN del virus del síndrome respiratorio y reproductivo porcino. Se trata de un método especifico y de alta sensibilidad para la detección del ARN vírico mediante la búsqueda del gen Nsp2. La amplificación se realizó en el termociclador Applied Biosystems™ Fast 7500 & 7500 Real-Time PCR y luego se siguió la metodología descrita en Cotaquispe et al.7.
Retrotranscripción convencional (RT-PCR) in house del gen ORF5Transcripción reversa (ADNc)El material genético viral extraído fue sometido a una retrotranscripción para obtener ADNc utilizando un termociclador Biometra TOne (Analytik Jena), siguiendo las instrucciones del Kit-OneScript® Hot cDNA Synthesis ABM®. Luego se siguió la metodología descrita en Cotaquispe et al.7.
PCR convencional del gen ORF5Los ADNc fueron amplificados en el termociclador antes mencionado. La reacción tuvo un volumen final de 20μl, con 3μl de ADNc a 40ng/μl, 10μl de mezcla maestra de 2X PCR HotStart ABM®, 5μl de agua libre de nucleasas (ABM®) y 1μl de cada cebador: SKA-F-5́GGTGGGCAACKGTTTTAGCCTGTC-3́, SKA-R-5́GGTAATAGARAAYGCCAAAAGCAC C-3́, a 10pmol/μl. Se emplearon los cebadores para el gen ORF5 que codifica la GP5 descritos en Key et al.11, los que fueron validados según los parámetros de sensibilidad, especificidad, límite de detección, repetitividad y robustez. Las condiciones de amplificación fueron un ciclo desnaturalización inicial a 94°C durante 5min seguido de 45 ciclos de desnaturalización a 94°C por 45s, alineamiento a 60°C por 60s y extensión a 72°C por 45s, con una extensión final a 72°C por 10min. El producto final fue un fragmento de 723pb que flanquea las regiones entre 13.400 y 14.000kb.
Electroforesis, purificación y secuenciaciónLos productos amplificados fueron revelados mediante cámara de electroforesis horizontal CS Cleaver Scientific multiSUB™ Choice con geles de agarosa de grado molecular SCL AG500 Cleaver Scientific al 1%, utilizando Fluorescent Dye-DNA/Safe-Green™ ABM® y el Ladder 100 bp Opti-DNA Marker 50 bp-1.5kb ABM®. Los productos se visualizaron en un fotodocumentador UVP UVsolo touch de Analytik Jena. Luego, los trozos del gel con el amplificado del tamaño esperado fueron concentrados con el kit de purificación de ácidos nucleicos innuPREP (Analytik Jena). Los productos purificados fueron secuenciados por ambas hebras con el servicio provisto por la empresa Macrogen (Corea) mediante el BigDye v3.1 sequencing terminator kit (Life Technologies) y un analizador genético ABI 3137 XL (Life Technologies).
Análisis filogenético del gen ORF5Los cromatogramas secuenciados se analizaron utilizando el paquete de secuencia Chromas Lite® v.2.6.6 y unieron a las 58 secuencias de referencia del GenBank: 9 de ellos se correspondieron con cepas peruanas (MH791386.1; MH791385.1; MH791383.1; MH791388.1; MH791379.1; MH791378.1; MH791376.1; MH791391.1; MH791390.1) y el resto con cepas patogénicas aisladas del resto del mundo (MF326985.1; DQ009647.1; DQ306879.1; DQ306878.1; DQ306877.1; GQ857656.1; FJ349261.1; AY588319.1; DQ489311.1; GQ461593.1; EF484033.1; AY032626.1; EF635006.1; KF611905.1; KF287140.1; JQ715697.1; EU939312.1; GQ374442.1; JQ663556.1; HQ315837.1; FJ889130.1; JX512910.2; JQ663545.1; JN411262.1; AY743931.1; JF802085.1; GU047345.1; EU076704.1; AY366525.1; KC862567.1; AF325691.1; KC862577.1; DQ473474.1; AB288356.1; JN660150.1; EU807840.1; EU262603.1; AF184212.1; DQ176019.1; AF331831.1; AF176348.2; U87392.3; MF279253.1; EF536003.1; AF066183.4; DQ988080.1; AY424271.1; AF494042.1; EF112445.1.).
Se efectuó el estudio filogenético molecular por el método de máxima verosimilitud mediante el análisis de sustitución nucleotídica, utilizando el modelo de parámetro Tamura 3. Los valores de robustez del árbol en los que los taxones asociados fueron agruparon, se muestra en la parte superior de las ramas. Los valores de bootstrapping se calcularon utilizando 1.000 réplicas de alineación; para la búsqueda heurística se obtuvieron aplicando el método de Neighbor-Joining a una matriz de distancias por pares estimadas utilizando el enfoque de máxima verosimilitud compuesta (MCL). El árbol está dibujado a escala, con las longitudes de las ramas medidas en número de sustituciones por sitio. El análisis involucró 82 secuencias de nucleótidos. Las posiciones de codón incluidas fueron 1ª+2ª+3ª+sin codificación. Hubo un total de 603 posiciones en el conjunto final de datos. Los análisis evolutivos se realizaron en MEGA® 625 y DNAMAN® v.10. En el análisis de homología de nucleótidos y aminoácidos se incluyeron las 58 variantes de referencia del linaje 1, 5 y 8 de las especies Betaarterivirus americense y Betaarterivirus europensis mediante el programa DNAMAN® versión 10.0, Lynnon Co28.
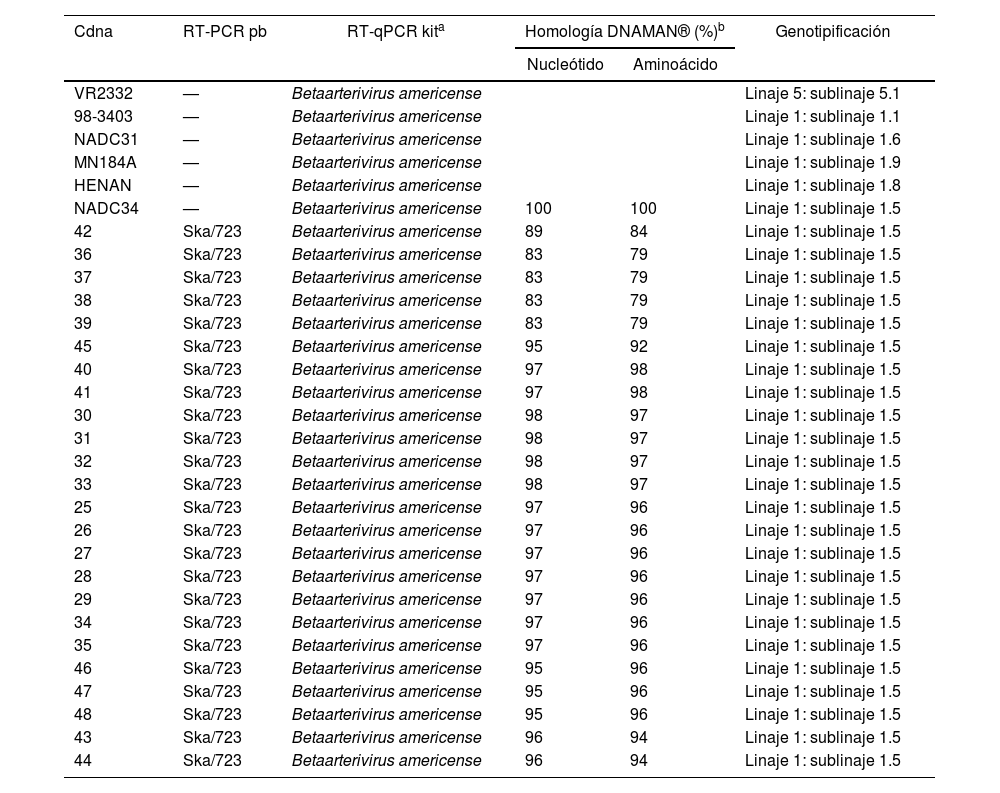
ResultadosLas muestras de suero se analizaron utilizando un kit comercial en búsqueda del gen Nsp2 del virus para la detección de las especies Betaarterivirus americense y Betaarterivirus europensis, y, en simultáneo, se desarrolló el procedimiento in house. Con ambos se obtuvo el mismo resultado. Identificamos 24 materiales genéticos de la especie Betaarterivirus americense con un Ct≤28 (tabla 1) y con bandas de 723 pb (cepas 25 a 48). Además, se ensayó y validó un kit in house con cebadores para el gen ORF5 de la GP5 del virus, usando los controles positivos de la vacuna inactivada de tipo 2 (americano) cepa Nebraska Prime Pac® PRRSV y de la vacuna inactivada tipo 1 (europeo) cepa VP-046 SUIPRABAC® PRRSV (fig. 1). Logramos la amplificación del gen, identificando 24 cepas positivas con tamaños específicos de 723pb con los cebadores SKAF y SKAR (fig. 1 y tabla 1). La secuenciación del total de los amplificados parciales demostró la presencia de los genes que sintetizan la glicoproteína 5, con longitudes de 603 nucleótidos y 201 aminoácidos funcionales.
Resultados de la identificación molecular de los 24 materiales genéticos del virus del síndrome reproductivo y respiratorio porcino (PRRSV) en muestras de sangre de cerdos de la región de Lima, Perú
| Cdna | RT-PCR pb | RT-qPCR kita | Homología DNAMAN® (%)b | Genotipificación | |
|---|---|---|---|---|---|
| Nucleótido | Aminoácido | ||||
| VR2332 | — | Betaarterivirus americense | Linaje 5: sublinaje 5.1 | ||
| 98-3403 | — | Betaarterivirus americense | Linaje 1: sublinaje 1.1 | ||
| NADC31 | — | Betaarterivirus americense | Linaje 1: sublinaje 1.6 | ||
| MN184A | — | Betaarterivirus americense | Linaje 1: sublinaje 1.9 | ||
| HENAN | — | Betaarterivirus americense | Linaje 1: sublinaje 1.8 | ||
| NADC34 | — | Betaarterivirus americense | 100 | 100 | Linaje 1: sublinaje 1.5 |
| 42 | Ska/723 | Betaarterivirus americense | 89 | 84 | Linaje 1: sublinaje 1.5 |
| 36 | Ska/723 | Betaarterivirus americense | 83 | 79 | Linaje 1: sublinaje 1.5 |
| 37 | Ska/723 | Betaarterivirus americense | 83 | 79 | Linaje 1: sublinaje 1.5 |
| 38 | Ska/723 | Betaarterivirus americense | 83 | 79 | Linaje 1: sublinaje 1.5 |
| 39 | Ska/723 | Betaarterivirus americense | 83 | 79 | Linaje 1: sublinaje 1.5 |
| 45 | Ska/723 | Betaarterivirus americense | 95 | 92 | Linaje 1: sublinaje 1.5 |
| 40 | Ska/723 | Betaarterivirus americense | 97 | 98 | Linaje 1: sublinaje 1.5 |
| 41 | Ska/723 | Betaarterivirus americense | 97 | 98 | Linaje 1: sublinaje 1.5 |
| 30 | Ska/723 | Betaarterivirus americense | 98 | 97 | Linaje 1: sublinaje 1.5 |
| 31 | Ska/723 | Betaarterivirus americense | 98 | 97 | Linaje 1: sublinaje 1.5 |
| 32 | Ska/723 | Betaarterivirus americense | 98 | 97 | Linaje 1: sublinaje 1.5 |
| 33 | Ska/723 | Betaarterivirus americense | 98 | 97 | Linaje 1: sublinaje 1.5 |
| 25 | Ska/723 | Betaarterivirus americense | 97 | 96 | Linaje 1: sublinaje 1.5 |
| 26 | Ska/723 | Betaarterivirus americense | 97 | 96 | Linaje 1: sublinaje 1.5 |
| 27 | Ska/723 | Betaarterivirus americense | 97 | 96 | Linaje 1: sublinaje 1.5 |
| 28 | Ska/723 | Betaarterivirus americense | 97 | 96 | Linaje 1: sublinaje 1.5 |
| 29 | Ska/723 | Betaarterivirus americense | 97 | 96 | Linaje 1: sublinaje 1.5 |
| 34 | Ska/723 | Betaarterivirus americense | 97 | 96 | Linaje 1: sublinaje 1.5 |
| 35 | Ska/723 | Betaarterivirus americense | 97 | 96 | Linaje 1: sublinaje 1.5 |
| 46 | Ska/723 | Betaarterivirus americense | 95 | 96 | Linaje 1: sublinaje 1.5 |
| 47 | Ska/723 | Betaarterivirus americense | 95 | 96 | Linaje 1: sublinaje 1.5 |
| 48 | Ska/723 | Betaarterivirus americense | 95 | 96 | Linaje 1: sublinaje 1.5 |
| 43 | Ska/723 | Betaarterivirus americense | 96 | 94 | Linaje 1: sublinaje 1.5 |
| 44 | Ska/723 | Betaarterivirus americense | 96 | 94 | Linaje 1: sublinaje 1.5 |
: ADNc muestra positiva Montana cepa 25; Línea 4 (GP5-SKA): ADNc muestra positiva Montana cepa 26. Línea 7 (GP5-SKA): control positivo de la cepa Nebraska Prime Pac® de PRRSV. Líneas 2, 5 y 8 (bronquitis aviar): controles negativos de ADNc de IBV. Líneas 3, 6 y 9: blancos de PCR. Línea M (ladder): 100bp Plus Opti-DNA Marker ABM®.")
RT-PCR convencional in house de los genes ORF5 diseñados en laboratorio. Línea 1 (GP5-SKA): ADNc muestra positiva Montana cepa 25; Línea 4 (GP5-SKA): ADNc muestra positiva Montana cepa 26. Línea 7 (GP5-SKA): control positivo de la cepa Nebraska Prime Pac® de PRRSV. Líneas 2, 5 y 8 (bronquitis aviar): controles negativos de ADNc de IBV. Líneas 3, 6 y 9: blancos de PCR. Línea M (ladder): 100bp Plus Opti-DNA Marker ABM®.
Las genealogías construidas a partir del gen ORF5 mediante sustitución nucleotídica utilizando máxima verisimilitud se muestran en la figura 2. La inferencia filogenética fue estimada empleando un alineamiento múltiple de secuencias de la especie Betaarterivirus europensis con sus variantes el linaje 1, sublinajes 1.A y 1.C (violeta), y el linaje 3 (gris). Lo mismo se efectuó con la especie Betaarterivirus americense con sus variantes del linaje 1: sublinajes 1.1, 1.5, 1.6, 1.8 y 1.9, linaje 5 con la variante representativa el sublinaje 5.1 -VR2332 y el linaje 8 con las variantes representativas CH-1a o CH-1R, añadiendo las cepas de la investigación y las cepas peruanas de referencia ORF5 (fig. 2).
 aplicado al gen ORF5 de las 24 cepas, con un bootstrapping de 1.000 réplicas de alineación. Los análisis evolutivos se realizaron en MEGA® v.6 y DNAMAN® v.10.")
Árbol filogenético de las especies Betaarterivirus europensis y Betaarterivirus americense. Se utilizó sustitución nucleotídica mediante máxima verosimilitud con el método de Neighbor-Joining, enfoque de máxima verosimilitud compuesta (MCL) aplicado al gen ORF5 de las 24 cepas, con un bootstrapping de 1.000 réplicas de alineación. Los análisis evolutivos se realizaron en MEGA® v.6 y DNAMAN® v.10.
La genealogía mostró valores bootstrapping cercanos al 100% para un árbol ramificado, dando como resultado dos especies de PRRS. Sin embargo, los clados que agruparon a los tres linajes muestran valores altos de robustez, evidenciando una relación clara para la inferencia parental dentro de la especie Betaarterivirus americense. Nuestros 24 materiales genéticos virales se distinguieron en el linaje 1, sublinaje 1.5, según las ramificaciones internas, con valores de bootstrapping del 100%. Los valores de confianza en el linaje 1 formado por los materiales genéticos del estudio y los materiales genéticos peruanos de referencia permitieron demostrar la tipificación relacionada específicamente con el sublinaje 1.5 de la variante NADC34 (verde), acompañado por el sublinaje 1.6 NADC31 (verde). Estas variantes tienen importancia porque han sido reportadas anteriormente y es un hecho preocupante su gran variabilidad. Los sublinajes que acompañan nuestros hallazgos son el 1.1, 1.8 y 1.9 (celeste). También el linaje 5 con el sublinaje 5.1 de la variante VR2332 y el linaje 8 con los sublinajes 8.7 y 8.9 (bordó) (fig. 2).
El origen evolutivo de nuestras cepas en relación con la variante NADC34 fue estudiado al alinear los 603bp del gen ORF5 y observar el árbol filogenético. Su relación con otras variantes del mismo linaje confirmó su clasificación y tipificación, por lo tanto, nos permitió tener el primer reporte completo de las cepas peruanas en el linaje 1 y sublinaje 1.5 de la especie Betaarterivirus americense. Asimismo, se observó la alta divergencia de las cepas 36, 37, 38, 39 y 42, mostrando una amplia distancia genética en la rama al formar subgrupos genéticos que se diferencian de las otras cepas.
El análisis de homología demostró que los 24 materiales genéticos del gen ORF5 tuvieron altos y bajos porcentajes de identidad nucleotídica y aminoacídica, lo que significa una marcada variabilidad genética con la cepa NADC34, con la que se alineó. Las cepas 25 al 35, 40, 41 y 43 al 48 obtuvieron valores altos de homología nucleotídica, del 95-98%, y aminoacídica, del 92-98%. Por otra parte, las cepas 36, 37, 38 y 39 mostraron una alta variabilidad genética con valores bajos de homología nucleotídica, del 83%, y aminoacídica, del 79%. La cepa 42 tuvo una homología nucleotídica del 89% y aminoacídica del 84% en relación con la variante NADC34 del sublinaje 1.5 en un análisis comparativo de las 58 secuencias más representativas y patógenas (fig. 3).
 aplicado al gen ORF5 de las 24 cepas, con un bootstrapping de 1000 réplicas de alineación, mediante el programa DNAMAN® v.10.")
Árbol filogenético de las especies Betaarterivirus europensis PRRSV-1 y Betaarterivirus americense PRRSV-2. Se empleó homología nucleotídica, enfoque de máxima verosimilitud compuesta (MCL) aplicado al gen ORF5 de las 24 cepas, con un bootstrapping de 1000 réplicas de alineación, mediante el programa DNAMAN® v.10.
Las especies del PRRSV tienen una capacidad epidémica alta y constituyen una amenaza para la economía de la industria porcina18. Se han realizado innumerables esfuerzos para controlar el virus, pero las vacunas aún no garantizan una protección satisfactoria debido a la capacidad mutagénica y la aparición de nuevas variantes de las especies Betaarterivirus europensis y Betaarterivirus americense; la eficiencia de aquellas cae drásticamente frente a confrontaciones heterólogas, aumentando los brotes locales y causando nuevas epidemias de la enfermedad11,12.
En el Perú, el virus se detectó a finales de la década que se inició en 1990 y desde entonces se han incrementado los reportes tardíos de pequeñas epidemias en diversas regiones de Lima19. Sin embargo, la escasa vigilancia epidemiológica y la falta de tecnología para detectar rápidamente las nuevas variantes están causando grandes pérdidas económicas en las granjas afectadas20.
El gen ORF5, que codifica la glicoproteína 5, es ampliamente utilizado para la investigación epidemiológica molecular, así como para evaluar la virulencia y la variabilidad. Las aplicaciones del análisis filogenético basado en este gen son muy similares a las del genoma completo según los antecedentes bibliográficos, ya que tiene una alta tasa de mutación y recombinación y, por lo tanto, se emplea en secuenciación para encontrar estas variaciones.
Los resultados de los diagnósticos rápidos evaluados en este estudio, en los que se empleó una porción del genoma viral y técnicas sensibles, revelaron la presencia de 24 cepas de la especie Betaarterivirus americense en la región de Lima; estos datos se asemejan a los reportados por Ramírez et al.19,20, quienes mostraron variantes del linaje 1 patogénicas en la región.
La inferencia filogenética realizada a partir de las secuencias extraídas de la base de datos, que fueron elegidas por ser responsables de graves brotes epidémicos, muestra dos ramas pertenecientes a las especies del virus, los linajes 1, 5 y 8 (fig. 2), e indica la buena relación filogenética de los aislados utilizando los primers empleados por Key et al.11. La genealogía también mostró que las cepas evaluadas en esta investigación mediante tipificación molecular pertenecen al linaje 1, estos mismos datos fueron reportados por Ramos et al.21, pero nuestro objetivo fue categorizar los sublinajes presentes en la región de Lima y los resultados muestran la presencia del sublinaje 1.5 con la cepa representativa NADC34 utilizando el gen ORF5; esta información y clasificación fueron validadas por Nguyen et al.16,26.
Los aislados peruanos de referencia fueron reportados por Ramírez et al.20 y muestran la relación con la variante NADC34, estos fueron tipificados como 1-7-4 en su mayoría utilizando RFLP10,25. En una publicación previa7 nuestro grupo ha confirmado estos resultados; la investigación basada en el gen ORF7 puso de manifiesto la presencia de dos linajes: el linaje 1 (sublinaje 1.5) y el linaje 5 (sublinaje 5.1) en un muestreo realizado en el año 2018. Entonces estos datos previos ya daban cuenta de la prevalencia y presencia del linaje 1, sublinaje 1.5, pero en esta segunda investigación muestreada en el año 2019 no hubo presencia del linaje 5 (sublinaje 5.1). Sin duda, es importante hacer un muestreo más exhaustivo para evidenciar con mayor amplitud y dar seguimiento al linaje 5 (sublinaje 5.1).
El análisis filogenético mostró la realidad de la rápida evolución y dispersión del sublinaje 1.5 del virus; nos proponemos estudiar más a fondo las características moleculares de la variabilidad viral en futuras investigaciones.
FinanciaciónEste trabajo fue ejecutado y financiado en las instalaciones de la Unidad de Negocio Nutrición y Salud Animal; Área de Innovación y Desarrollo, Corporación Montana S.A.
Conflicto de interesesLos autores no tenemos ningún tipo de conflicto de intereses al presentar el presente artículo para su publicación.
El trabajo de investigación se desarrolló en las instalaciones del Centro Investigación de la Unidad de Negocio de Nutrición y Salud Animal; Área de Innovación y Desarrollo, Corporación Montana S.A. por ello se agradece a la empresa y al Dr. Nicolás Sitaras por el apoyo al desarrollo de este trabajo.
