










Los inhibidores de la tirosina cinasa son utilizados para el tratamiento de diversas neoplasias interfiriendo en múltiples vías de proliferación celular y angiogénesis tumoral. Estos fármacos presentan efectos adversos de clase, destacándose entre ellos la afectación de la función tiroidea. Existen diferentes mecanismos propuestos por los cuales estos agentes orales llevan tanto al hipotiroidismo como a la tirotoxicosis. Aún no existe consenso sobre el seguimiento y tratamiento ante la aparición de estos efectos.
Tyrosine kinase inhibitors are used for the treatment of different types of tumours, interfering in various cell proliferation pathways, and tumour angiogenesis. These oral agents have side effects, thyroid dysfunction outstanding among them. There are different mechanisms through which these agents lead to hypothyroidism, as well as thyrotoxicosis. There is still no consensus on the treatment and follow-up of the above mentioned effects.
Los receptores de tirosina cinasa (TC) son reguladores centrales de vías de proliferación, diferenciación y apoptosis celular. La disregulación de la señalización mediada por estas proteínas se encuentra involucrada en la patogénesis de múltiples neoplasias malignas.
Los inhibidores de TC (ITC) constituyen un grupo de pequeñas moléculas de administración oral diseñadas para interferir en las vías oncogénicas dependientes de TC. Ofrecen una alta ventana terapéutica con menor toxicidad en comparación con la quimioterapia convencional1.
En pacientes con cáncer, las anormalidades de la función tiroidea pueden estar variablemente asociadas con el tumor en sí mismo, con la realización de procedimientos diagnósticos que utilizan contrastes iodados o con el tratamiento instaurado para dicha neoplasia2.
A medida que ha avanzado la experiencia con el uso de los ITC se han descripto varios efectos adversos, entre los cuales se destaca la alteración de la función tiroidea1,3.
Es objetivo de esta monografía poder actualizar cuál es el mecanismo de acción de estos fármacos, cuáles son sus usos, de qué manera y qué tipo de alteración de la función tiroidea producen y, por ende, cuál sería el control apropiado por parte del endocrinólogo tratante cuando el paciente reciba dicho tratamiento o se plantee la posibilidad de iniciarlo por parte de los oncólogos.
Tirosina cinasaFunción y clasificaciónLas TC son enzimas que catalizan la transferencia de fosfatos del adenosín trifosfato (ATP) a determinadas proteínas y desempeñan un papel central en la modulación de señales de crecimiento celular interviniendo en diversos procesos normales de regulación. Formas activas de estas enzimas pueden causar aumentos de la proliferación de células tumorales, generar efectos antiapoptóticos así como, promover la angiogénesis y el desarrollo de metástasis. Por tal motivo constituyen un blanco especial e importante de las terapias dirigidas en oncología. Pueden ser clasificadas en 2 grandes grupos: receptores de TC y no receptores de TC4.
Receptores de tirosina cinasaLos receptores de TC median la acción biológica de una amplia variedad de ligandos en gran cantidad de tipos celulares. Todos presentan un dominio extracelular de unión al ligando, un dominio transmembrana y otro intracelular que incluye el dominio catalítico TC al que se unen el ATP y sustratos proteicos. Estas proteínas median las acciones biológicas de diversos ligandos entre ellos: insulina, factor de crecimiento epidérmico (EGF), factor de crecimiento derivado de plaquetas (PDGF) y factor de crecimiento derivado de la célula endotelial vascular (VEGF), entre otros5 (fig. 1).

Estructura de los receptores de TC.
Adaptado de Lodish y Stratakis1.
Los receptores de TC se encuentran como monómeros. Cuando el ligando se une al dominio extracelular del receptor se dimeriza. Como consecuencia de ello, el receptor se autofosforila y en este estado el receptor es capaz de fosforilar a otras proteínas. En cuanto a los mecanismos que intervendrían en la finalización de la acción hormonal, se describe que los mismos estarían mediados por fosfatasas, así como por procesos de endocitosis mediada por el receptor y por la fosforilación por serina-treonina cinasas5.
Inhibidores de tirosina cinasaMecanismo de acciónExisten diferentes mecanismos por los cuales la alteración de la regulación de los receptores de TC interviene en la oncogénesis3. El primero de ellos consiste en un rearreglo cromosómico, que determina una activación constitutiva del receptor en ausencia de ligando. El ejemplo de ello es la formación de la oncoproteína BCR-ABL o cromosoma Philadelphia en leucemia mieloide crónica (LMC). Otra forma de disregulación es la mutación de la cinasa (dominio catalítico) del receptor, como se ve en la leucemia mieloide aguda por la mutación del TC 3 tipo Fms de hígado fetal (FLT3) o en el receptor del EGF en cáncer de pulmón no de células pequeñas. Un tercer mecanismo consistiría en la expresión aumentada o aberrante del receptor, de su ligando o de ambos. Finalmente, la ausencia de factores que limiten la actividad TC (fosfatasas) sería otro de los mecanismos propuestos. La mutación de las TC aumenta la supervivencia, proliferación y resistencia a fármacos de las células neoplásicas, pudiendo estimular la angiogénesis tumoral, así como su invasividad y potencial metastásico4.
Los ITC comprenden un conjunto de pequeñas moléculas de administración oral que previenen la fosforilación de diversos receptores de TC compitiendo con la unión del ATP al dominio catalítico de los mismos, dado que comparten con este similitud estructural6.
Dado que el dominio catalítico de estos receptores se encuentra altamente conservado, varios ITC pueden desarrollar su efecto inhibitorio sobre un gran espectro de cinasas afectando de este modo múltiples vías de señalización1.
UsosLos ITC son utilizados para una gran variedad de enfermedades malignas que incluyen el tratamiento de tumores sólidos y hematológicos entre ellos LMC, tumores estromales gastrointestinales (GIST) recurrentes o metastásicos, carcinoma células renales (CCR) recurrentes o metastásicos, hepatocarcinoma, carcinoma pulmonar no de células pequeñas, tumores neuroendocrinos y tiroideos tanto diferenciados (CDT) avanzados como medulares.
Cada uno de estos fármacos tiene actividad sobre determinadas TC, siendo esto lo que determina su efectividad en el control de ciertas neoplasias y no en otras1 (tabla 1).
Usos y blancos terapéuticos de los principales ITC
| Fármaco | Usos | Blanco terapéutico |
|---|---|---|
| Imatinib | LMC. LLA. Cromosoma Philadelfia positivo (LLA). GIST | BCR-ABL, KIT, PDGFRA, CSFR |
| Dasatinib | LMC y LLA cromosoma Philadelfia positivo. | BCR-ABL, KIT, PDGFRA, Familia de cinasas SRC |
| Nilotinib | LMC | BCR-ABL, KIT, PDGFR A |
| Sunitinib | GIST, CCR, tumores pancreáticos neuroendocrinos | VEGFR 1-3, KIT, FLT3, PDGFR A y B, RET |
| Sorafenib | CCR, hepatocarcinoma, CDT | VEGFR2, PDGFR, FLT3, RET, KIT, BRAF |
| Vandetanib | Mieloma, gliomas, carcinoma pulmonar no de células pequeñas, carcinoma medular avanzado | VEGFR, RET, EGFR |
| Motesanib | GIST, cáncer de mama, tumores neuroendocrinos, tiroideos, carcinoma colorrectal, carcinoma pulmonar no de células pequeñas | VEGFR 1-3, PDGFR, KIT, RET |
| Axitinib | CCR, carcinoma pulmonar no de células pequeñas, melanoma, tumores tiroideos, pancreáticos, carcinoma mama | VEGFR 1-3 |
| Pazopanib | CCR | VEGFR 1-3, KIT, PDGFR |
BRAF: oncongén homólogo al sarcoma viral murino; B1KIT: oncogén homólogo al sarcoma viral felino v-kit; CDT: carcinoma diferenciado de tiroides; CSFR: Receptor del factor de crecimiento de células madre; EFGR: receptor del factor de crecimiento epidérmico; LLA: leucemia linfoblástica aguda; LMC: leucemia eosinofílica crónica; PDGFR: receptor del factor de crecimiento derivado de plaquetas; RET: rearreglado durante la transferencia; VEGFR: receptor del factor de crecimiento endotelial vascular.
Si bien son raramente letales pueden conducir a diversas comorbilidades8. Se han demostrado sobre corazón, pulmones, hígado, riñones, piel, coagulación y sistema endocrino.
Teniendo en cuenta que las TC están tan ampliamente distribuidas y cumplen diversos papeles fisiológicos en distintos órganos y sistemas, es esperable que su inhibición no solo logre el efecto deseado (su eficacia), sino efectos indeseados (tóxicos)3.
Los ITC desarrollan efectos adversos de clase (comunes a todos los inhibidores) con un perfil de toxicidad similar en todas las patologías. Cada inhibidor presenta una distinta incidencia de los diversos tipos de reacciones adversas y algunos hasta desarrollan efectos propios8.
En relación con el sistema endocrino estos fármacos afectan sobre todo a la función tiroidea, mientras que el resto de los ejes se ven raramente comprometidos.
Disfunción tiroidea inducida por inhibidores de tirosina cinasaLos ITC pueden afectar a la función tiroidea mediante distintos mecanismos fisiopatológicos que afectan al tejido tiroideo o el metabolismo de hormonas tiroideas9.
Causan hipotiroidismo o hipertiroidismo de novo o agravan un hipotiroidismo preexistente. La principal alteración observada es el hipotiroidismo primario (clínico 32-85% y subclínico en casi el 100% de los casos). También se describió asociación con descensos del valor de tirotrofina (TSH) o tirotoxicosis (transitoria 0-24%, persistente 0-5%)2.
El inicio de la disfunción tiroidea inducida por ITC es variable entre las 4 y 94 semanas desde el comienzo del tratamiento, siendo el promedio las 4 semanas en estudios prospectivos2.
HipotiroidismoEl hipotiroidismo es la patología endocrina más frecuentemente observada secundaria al uso de ITC, siendo reversible en la mayoría de los pacientes pero no en todos3.
La aparición de fatiga con el uso de estos fármacos fue lo que motivó la profundización en la investigación a fin de conocer si esta se trataba de un efecto propio de los ITC o estaba vinculada a disfunción de la glándula tiroidea10.
El primer estudio en el que se constató disfunción tiroidea por el uso de ITC fue realizado por De Groot et al. en 8 pacientes tiroidectomizados por carcinoma medular y tratados con imatinib, que desarrollaron elevaciones marcadas de TSH mientras que T4 libre y totales se mantuvieron en el rango de referencia. A pesar de duplicar la dosis de levotiroxina, la función tiroidea solo logró ser normalizada en 3 de los 8 pacientes evaluados11.
Posteriores a este trabajo aparecieron múltiples publicaciones sobre disfunción tiroidea en pacientes no tiroidectomizados (tabla 2).
Frecuencia de hipotiroidismo reportado mediada por diferentes ITC
| ITC | Frecuencia de hipotiroidismo |
|---|---|
| Sunitinib | 53-85% |
| Sorafenib | 20-36% |
| Imatinib | 90-100% (solo en pacientes tiroidectomizados) |
| Motesanib | 22% |
| Vandetanib | 89% |
| Axitinib | 83-92% (pequeño número de pacientes analizados) |
| Pazopanib | 10-29% |
| Tivozanib | 5% |
Adaptado de Ahmadieh y Salti7.
Como se ha descripto, el sunitinib fue aprobado para el tratamiento del CCR, GIST y tumores pancreáticos neuroendocrinos irresecables. Se administra por vía oral en ciclos de 6 semanas, 4 en las que el paciente recibe el fármaco «período on» seguidas por 2 de descanso «período off». Una de las primeras descripciones de disfunción tiroidea mediada por sunitinib fue realizada en 2006 por Desai et al. en pacientes con GIST resistentes a imatinib. Fueron evaluados 42 pacientes eutiroideos que recibieron sunitinib por al menos 3 ciclos completos. El 62% de los mismos desarrolló disfunción tiroidea. El riesgo de hipotiroidismo se vio incrementado cuanto mayor era el tiempo de tratamiento10.
En la literatura el sunitinib es el ITC que con mayor frecuencia produce hipotiroidismo (en estudios retrospectivos en el 53-85% de los pacientes analizados y en prospectivos en el 21-71%); ¿cuál podría ser la explicación de este fenómeno?12.
Fisiopatología: hipótesis propuestasDisminución de la vascularizaciónLa glándula tiroidea presenta la más elevada tasa de flujo sanguíneo por unidad de peso en comparación con cualquier otro tejido del organismo. Su vascularización depende fundamentalmente de la vía de señalización mediada por VEGFR12.
Existen 3 tipos de VEGFR denominados 1, 2 y 3 estando los 2 primeros involucrados en el proceso de angiogénesis y el tercero en la linfagénesis. Estas proteínas presentan varios ligandos entre ellos VEGFA, B, C, D y el factor de crecimiento placentario (PLGF). El mayor inductor de la angiogénesis es el VEGFA, que hace efecto principalmente sobre el VEGFR2. La unión de este ligando con el VEGFR1 es 10 veces menor en comparación a su afinidad por el VEGFR2. En la mayoría de los diferentes tipos tumorales se expresa esta vía de cinasas13 (fig. 2).

Vías de señalización involucradas en la vascularización tiroidea.
Adaptado de Makita y Liri12.
A nivel tiroideo, la unión de TSH con su receptor estimula la liberación por las células foliculares tiroideas de VEGFA que al unirse con el VEGFR2 estimula la proliferación de células endoteliales y su fusión, aumentando el tamaño de la luz vascular. De este proceso también participa como agente paracrino el PLGF uniéndose al VEFGR112.
El sunitinib, al inhibir esta vía angiogénica, provocaría hipotiroidismo por reducción del flujo vascular mediante la constricción y la regresión de capilares. Esto generaría isquemia a nivel glandular provocando disfunción tiroidea. Si el proceso isquémico fuera severo llevaría a la apoptosis del tirocito y a la destrucción del tejido. Por ende, sería el mismo mecanismo fisiopatológico el que ocasionaría hipotiroidismo y tiroiditis destructiva14.
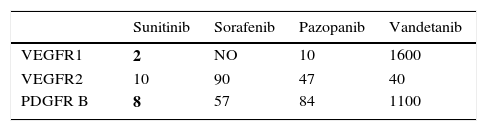
Otros ITC actúan por la vía del VEGFR, tales como el sorafenib, axitinib y pazopanib. Entonces, ¿por qué con sunitinib hay mayor frecuencia de evolución al hipotiroidismo? No solo la inhibición del VEGFR2 y VEGFR1 serían necesarias para generar isquemia tiroidea, sino también la del PDGFR. Esto es lo que diferencia al sunitinib del resto de los fármacos, dado que es el que presenta mayor afinidad por el VEGFR1 y el PDGFR12 (tabla 3).
Concentración inhibitoria del 50% de algunos ITC para diferentes receptores
| Sunitinib | Sorafenib | Pazopanib | Vandetanib | |
|---|---|---|---|---|
| VEGFR1 | 2 | NO | 10 | 1600 |
| VEGFR2 | 10 | 90 | 47 | 40 |
| PDGFR B | 8 | 57 | 84 | 1100 |
Los valores de concentración inhibitoria están expresados en nanomoles. En el texto se explica lo que diferencia al sunitinib del resto de los ITC en el sentido que este presenta mayor afinidad sobre VEGFR1 y PDGFR B que el resto.
Adaptado de Makita y Liri12.
En un reporte de casos se describe a una paciente de 60 años en tratamiento con sunitinib por un CCR metastásico que evoluciona a un hipotiroidismo clínico. Durante el «período on» del fármaco, la paciente desarrolla atrofia glandular y disminución de la vascularización tiroidea, revirtiendo durante el período de discontinuación del tratamiento14.
Atrofia glandularDesde el trabajo de Desai, donde se describen 2 pacientes que presentaron atrofia tiroidea secundaria probablemente a tiroiditis destructiva, se plantea este mecanismo como probable etiología de disfunción tiroidea1.
En un estudio de Shinohara et al. se evaluó a 17 pacientes con CCR metastásico a los que se les realizó tomografía computada con reconstrucción de imagen tridimensional previo a sunitinib, en el primer ciclo de tratamiento y luego de la suspensión del mismo. Los pacientes fueron seguidos durante un promedio de 6 ciclos y se observó una tasa media de reducción del volumen tiroideo del 30% e hipotiroidismo en la mitad de los mismos. El grupo clasificado como de alta reducción, con una disminución del volumen tiroideo mayor al 50% (n=8 pacientes), fue en el que se evidenció mayor incidencia de hipotiroidismo y el que recibió mayor tiempo de tratamiento con sunitinib. De estos 8 pacientes, 4 presentaron TSH suprimidas previas a la instalación del hipotiroidismo lo que conduciría a plantear a la tiroiditis destructiva como posible mecanismo de atrofia tiroidea15.
De los pacientes a los que se les discontinuó el sunitinib (n=4) ninguno presentó recuperación del volumen tiroideo. Si bien el tiempo de seguimiento postratamiento fue corto, esto sugeriría la posibilidad de un daño irreversible de la glándula causado por este fármaco15.
En un trabajo retrospectivo que incluyó a 42 pacientes con CCR tratados con sunitinib y sorafenib se analizaron la función tiroidea y los cambios de tamaño glandular por tomografía computada basalmente y a los 3, 6, 9 y 12 meses de seguimiento. Se observó que 21 pacientes desarrollaron hipotiroidismo y en ellos hubo una reducción del tamaño tiroideo que alcanzó el 68±21% a los 12 meses de tratamiento, siendo esta más pronunciada en los que recibieron sunitinib. En los pacientes que se mantuvieron eutiroideos no se observó reducción glandular16.
Rogiers et al. reportaron resultados similares en 2 pacientes con bocios nodulares en tratamiento con sunitinib por CCR, que requirieron hormona tiroidea aun después de suspendido el ITC17.
Inhibición en la captación de iodoEn el estudio prospectivo de Mannavola et al. se analizó a 24 pacientes con diagnóstico de GIST pero solo a 6 de ellos se les realizó captación y centellograma con 123I hacia el final de los «períodos on» y «off». Se observó una disminución en la captación durante las 4 semanas de tratamiento, con una normalización parcial o completa hacia el «período off». De estos 6 pacientes, solo 3 presentaron hipotiroidismo y los restantes elevaciones mínimas de tirotrofina. Los autores plantearon que la inhibición del simportador sodio/iodo (NIS) sería un mecanismo probable de desarrollo de hipotiroidismo mediado por sunitinib. Esto estaría dado por el flúor, que es un componente de su estructura y que actuaría como un inhibidor competitivo de este transportador. No obstante, el dosaje de flúor urinario fue normal en todos los pacientes, lo que no avalaría esta hipótesis18.
Sin embargo, en una publicación del 2008 se vio que incubando sunitinib a diferentes concentraciones en células tiroideas de rata (FRTL-5), la captación tiroidea de 125I aumentaba en forma dosis dependiente y el flujo de salida del mismo no se veía alterado por el agregado del fármaco. En este estudio in vitro la expresión del NIS medida por reacción en cadena de la polimerasa con transcriptasa reversa no se vio incrementada por el uso de sunitinib y tampoco se vio alteración en el receptor de TSH que regula la acción de este transportador. Este trabajo, en oposición a lo expuesto por Mannavola et al., plantea como probable etiología de hipotiroidismo una alteración a nivel de la organificación de iodo o en otro paso de la biosíntesis de hormonas tiroideas19.
Inhibición de la tiroperoxidasaWong et al. realizaron un estudio in vitro midiendo la actividad de la lactoperoxidasa, una enzima similar a la tiroperoxidasa. La potencia antitiroidea de sunitinib fue determinada por la concentración del fármaco capaz de inhibir el 50% de la actividad de la peroxidasa y se la comparó con la de las tionamidas. Se vio que la potencia antitiroidea de sunitinib fue un 25-30% menor respecto a la de propiltiouracilo. Dado que la glándula presenta una reserva hormonal de aproximadamente 2 meses, es de esperarse que el bloqueo parcial de la tiroperoxidasa mediado por sunitinib determine la aparición gradual de hipotiroidismo. Por otra parte, dado que la potencia inhibitoria de este fármaco es menor que la de las tionamidas, se explicaría por qué el sunitinib no generaría hipotiroidismo en todos los pacientes20 (fig. 3).

Potencia inhibitoria de sunitinib en comparación con tionamidas.
Adaptado de Wong et al.20.
Esta hipótesis se origina en la similitud estructural de las iodotironinas con los ITC y por ende se plantea que estos fármacos interferirían en el transporte de hormonas tiroideas. En 2 trabajos recientes realizados in vitro, se describe la inhibición no competitiva mediada por distintos ITC del transportador transmembrana monocarboxilado 8 (MCT 8) de hormonas tiroideas presente a nivel hipotálamo-hipofisario y en otros órganos. Esto reduciría el feedback hipotálamo-hipofisario-tiroideo ocasionando elevaciones de TSH e incrementos en los requerimientos de hormona tiroidea de pacientes tiroidectomizados21,22.
Aumento del metabolismo y requerimiento de hormona tiroidea¿Qué sucede en los pacientes con diagnóstico previo de hipotiroidismo que inician tratamiento con ITC? De Groot et al. observaron que los pacientes con tiroides in situ se mantuvieron eutiroideos con el uso de imatinib, mientras que los tiroidectomizados presentaron un aumento del requerimiento de levotiroxina de un promedio del 206%. Esto sugirió que la causa por la cual los ITC generaban aumentos en los requerimientos de hormona tiroidea estaba vinculada a un aumento del clearence de iodotironinas. En estos pacientes no hubo variaciones en los niveles de T4 total, libre y globulina ligadora de tiroxina, lo que descartó la posibilidad de que el ITC haya generado competencia en la unión de las hormonas tiroideas a su proteína transportadora o aumentos en los niveles de la misma. Tampoco esto estuvo dado por una inhibición del metabolismo dependiente de desiodinasas, dado que en este caso el nivel de T4 hubiera aumentado. Por tal motivo, los autores sugirieron un aumento del metabolismo de hormonas mediado por la conjugación con glucuronatos y sulfatos a nivel hepático. El imatinib en este caso actuaría como un inductor de las glucuroniltransferasas11.
Por su parte, el estudio de Dora et al., con una muestra muy pequeña y seguimiento a corto plazo, mostró que los pacientes con LMC tratados con imatinib no presentaron alteración de la función tiroidea. Los autores sugirieron que sería la interferencia en la absorción a nivel gastrointestinal de la levotiroxina mediada por este ITC lo que generaría disfunción tiroidea23.
En el año 2006, De Groot et al. publicaron el caso de una mujer de 73 años tiroidectomizada y ablacionada por un carcinoma folicular en tratamiento con levotiroxina. La paciente inició imatinib por un GIST metastásico que tuvo que ser discontinuado y reemplazado por sunitinib, y con ambos fármacos presentó un cuadro compatible con hipotiroidismo requiriendo un aumento de la dosis de levotiroxina hasta un total de 300 μg por día24.
Se ha descripto también que el aumento del metabolismo de hormonas tiroideas estaría provocado por un aumento en la actividad de la desiodinasa tipo 3. Kapper et al. realizaron un seguimiento tanto prospectivo (n = 15) como retrospectivo (n = 83) de pacientes en tratamiento con sunitinib por CCR o GIST metastásicos y analizaron en forma experimental la función tiroidea y la histología de ratas durante y luego del tratamiento con sunitinib. El 42% de los pacientes en el estudio retrospectivo desarrolló una elevación de TSH. Igual situación se evidenció en la rama prospectiva, donde también se observó un descenso de la relación T3/T3r. En ratas se evidenció una inducción de la desiodinasa 3 hecho que coincidió con el descenso de T3 y T4 en las mismas. Esta situación fue revertida luego de la suspensión del ITC. Se sabe que el factor 1 inducible por hipoxia (HIF-1) es un modulador positivo del gen de la desiodinasa 3. Si bien el sunitinib no aumenta los niveles de HIF-1 en forma directa al generar un medio con hipoxia (por su efecto antiangiogénico), generaría una up-regulation de este factor, lo que indirectamente llevaría a un incremento de la desiodinasa 325.
En un reciente estudio prospectivo sobre 21 pacientes tiroidectomizados por carcinoma tiroideo no medular tratados con levotiroxina a dosis supresivas y que recibieron sorafenib por 26 semanas, se observó un aumento en la dosis de hormona tiroidea ajustada por kilo de peso al final del estudio para mantener los valores de T4 libre. Los niveles de TSH fueron en aumento durante el seguimiento, mientras que los niveles de T3 se redujeron y los de T3 reversa aumentaron. Las relaciones T3/T4 y T3/T3r se redujeron significativamente reflejando un aumento en la actividad desiodinasa 326.
Disminución del clearence de tirotrofinaVerloop et al. analizaron a 20 pacientes tiroidectomizados y ablacionados por carcinoma tiroideo en tratamiento con levotiroxina a dosis supresivas. A ellos se les inyectó TSH recombinante antes y al final del uso del ITC (26 semanas). Se dosaron los niveles de TSH a las 48 y 96 h luego de la primera inyección y se calculó el área bajo la curva (ABC). Se planteó que dado que la dosis inyectada era la misma antes y al finalizar el tratamiento con sorafenib y que los niveles de TSH endógenos estaban suprimidos por el uso de levotiroxina, cualquier diferencia encontrada en los valores de TSH antes y después del tratamiento apuntarían hacia un efecto independiente del sorafenib sobre la misma. Se evidenció un aumento de los niveles de TSH a las 48 y 96 h, al igual que un aumento del ABC hacia el final del tratamiento respecto al inicio del mismo. Esto no estuvo influenciado por la TSH basal dado que la misma luego del sorafenib solo sufrió una leve modificación. Se postuló como conclusión que este ITC actuaría aumentando la vida media de TSH (aumentado su sialación), originando un menor metabolismo de la hormona27.
Autoinmunidad tiroideaEn la mayoría de los estudios de pacientes que presentaron hipotiroidismo inducido por ITC los anticuerpos antitiroperoxidasa, antitiroglobulina y antirreceptor de TSH se mantuvieron negativos, lo cual descartaría la etiología autoinmune. Sin embargo, en la citología de pacientes en tratamiento con sunitinib se ha descripto el diagnóstico de tiroiditis linfocitaria. Se propuso que serían las citocinas liberadas por los linfocitos T activos (interferón gamma, interleucina 2) las que mediarían la patogénesis de esta tiroiditis28.
Patrón de instalación de hipotiroidismoEn las diferentes publicaciones sobre disfunción tiroidea e ITC se han evidenciado oscilaciones del valor de TSH hasta que los pacientes llegan al estado de hipotiroidismo. Particularmente con sunitinib, se han descripto aumentos de tirotrofina en los «períodos on» y su normalización hacia los «periodos off» pero a medida que avanzan los ciclos de tratamiento la aparición del hipotiroidismo se vuelve permanente y el paciente requiere reposición con levotiroxina29.
En un estudio prospectivo observacional de pacientes con diagnóstico de CCR metastásico y GIST resistentes/intolerantes a imatinib y tratados con sunitinib, se analizó la función tiroidea basal y en los días 1 y 28 de cada ciclo de tratamiento. De 59 pacientes analizados, 20 fueron eutiroideos, 20 presentaron en una sola oportunidad una elevación de TSH sin requerir tratamiento, mientras que 16 desarrollaron hipotiroidismo clínico o subclínico, por lo que se les indicó levotiroxina. En 3 pacientes los niveles de TSH se encontraron por debajo del límite inferior con valores de hormonas periféricas normales. De los 16 pacientes que desarrollaron hipotiroidismo, 4 presentaron un patrón bioquímico previo de hipertiroidismo transitorio, cuadro compatible con tiroiditis. El tiempo medio para la aparición de valores alterados de TSH fue de 4 semanas variando en un rango entre 2 y 26. Como se mencionó previamente, las concentraciones de TSH presentaron una evolución en zigzag con niveles normales en el basal, elevándose al día 28 luego de 4 semanas de tratamiento con sunitinib y normalizándose en las 2 semanas de descanso. Los autores postularon que la alteración en la vascularización tiroidea observada con este ITC, con recuperación de la misma una vez suspendido el fármaco, podría explicar este patrón rítmico de secreción de TSH30 (fig. 4).

Patrón de instalación del hipotiroidismo.
Adaptado de Wolter et al.30.
En los pacientes que desarrollaron disfunción tiroidea, este patrón se observó durante la mayoría de ciclos de tratamiento. Fue justamente este grupo el que recibió mayor tiempo de administración de sunitinib (48 semanas vs. 21 semanas en los pacientes sin disfunción)30.
TratamientoAl igual que en la población general, se propone que pacientes con valores de TSH mayores o iguales a 10 mU/l deben ser tratados con hormona tiroidea. Sin embargo, existe controversia en aquellos con valores entre 4,5 y 10 mU/l. No existen a la fecha guías elaboradas por las diferentes sociedades sobre el manejo del hipotiroidismo inducido por ITC31.
En el trabajo de Wolter et al. se propuso un algoritmo de tratamiento de la disfunción tiroidea inducida por sunitinib. Los autores plantearon que pacientes con valores de TSH superiores a 10 mU/l persistentes y clínica de hipotiroidismo (aun con T4 dentro de rango de referencia) o T4 baja en el día 1 de un ciclo de sunitinib deberían ser medicados. Dado que los valores de TSH pueden disminuir e incluso normalizarse al final de las 2 semanas de descanso de sunitinib, la decisión de tratamiento se debería focalizar en el nivel de TSH del día 1 de un nuevo ciclo. No recomendaron tratar a pacientes con TSH elevada en el día 28, dado que como estos valores pueden normalizarse sin mediar intervención, esto conduciría a un sobretratamiento30.
Se ha visto que solo el 50-79% de los pacientes en tratamiento con ITC y con diagnóstico de disfunción tiroidea presentan mejoría sintomática de la fatiga con el uso de levotiroxina a pesar de la normalización de los niveles de TSH. Esto marcaría que la relación entre la fatiga y el hipotiroidismo inducida por estos fármacos sería solo especulativa32.
Por otra parte, se propone que aquellos pacientes hipotiroideos previos al inicio de ITC probablemente requieran aumentos de su dosis de levotiroxina2. El hipotiroidismo no es una indicación para suspender o reducir la dosis del ITC1.
Así mismo en pacientes tiroidectomizados bajo tratamiento con hormona tiroidea que deben ser medicados con imatinib, vandetanib, axitinib o motesanib se debería considerar duplicar la dosis de LT4 en el inicio de la terapia2.
Se ha observado que a pesar de la reposición correcta de levotiroxina, en algunos casos es difícil alcanzar el estado de eutiroidismo dadas las fluctuaciones en los valores de TSH y T433.
HipertiroidismoExisten reportes que parecerían indicar como fisiopatología de la tirotoxicosis por ITC un mecanismo símil a tiroiditis destructiva con hipertiroidismo seguido de hipotiroidismo34, así como otros casos de tiroiditis con evolución a hipotiroidismo definitivo luego de una fase de hiperfunción35.
En un reporte de van Doorn et al. se evidencia claramente la presencia de tiroiditis. En primer lugar describe a una paciente de 49 años con hepatocarcinoma irresecable que inició sorafenib 400mg 2 veces por día con exámenes de función tiroidea basales dentro de parámetros de normalidad. A 17 semanas de tratamiento se presentó asintomática y con valores de TSH suprimidos y T4 libre elevados, mientras que a las 64 semanas evidenció un cuadro clínico compatible con hipotiroidismo por el que requirió el inicio de levotiroxina. La paciente suspendió sorafenib a las 72 semanas de tratamiento por progresión de la enfermedad de base pero continuó con levotiroxina aun después de suspendido el ITC (fig. 5)36.

Tiroiditis en paciente en tratamiento con sorafenib.
Adaptado de van Doorn et al.36.
Por otra parte, se ha evidenciado la aparición de hipertiroidismo en pacientes con LLC en tratamiento con nilotinib y dasatinib37.
El tratamiento de la tirotoxicosis dependiente de ITC dependería de la gravedad del paciente, pudiendo adoptarse una conducta expectante en casos leves y requiriendo del uso de glucocorticoides y betabloqueantes en casos severos35.
También se describe en la literatura un caso de crisis tirotóxica inducido por sorafenib en una paciente con un CCR38.
SeguimientoLas distintas sociedades de endocrinología aún no han elaborado guías sobre el seguimiento de aquellos pacientes que desarrollan disfunción tiroidea inducida por ITC. La mayoría de los trabajos citan el algoritmo de manejo de tratamiento y el control de la disfunción tiroidea de pacientes bajo sunitinib elaborado por Wolter et al. en su estudio prospectivo observacional30 (fig. 6).

Algoritmo para el diagnóstico y tratamiento de la disfunción tiroidea inducida por sunitinib.
Adaptado de Wolter et al.30.
Como se ve, una elevación de TSH en el día 1 del ciclo indica más apropiadamente un daño clínico de la glándula tiroidea que requerirá tratamiento sustitutivo. En cambio, la determinación de TSH el día 28 del ciclo aumentaría las probabilidades de detectar precozmente la disfunción tiroidea subclínica o transitoria que no requerirá tratamiento. En lo que respecta a otros ITC se aconseja el análisis de TSH pre tratamiento seguido de una evaluación mensual y luego cada 2-3 meses. Se necesitan estudios prospectivos para definir en forma precisa cuando es el mejor momento para analizar la función tiroidea en pacientes bajo sunitinib y otros ITC2.
ConclusionesA medida que se fue confeccionando esta monografía, se han incrementado los artículos publicados en distintas revistas sobre ITC. Esto demuestra, por un lado, el interés creciente sobre lo que podríamos denominar nuevas terapias antineoplásicas y, por el otro, el aumento en el espectro de tumores que pueden ser tratados con el uso de estos fármacos, dado que cada vez es mayor el conocimiento sobre patogénesis molecular en el desarrollo tumoral39,40.
Analizar la toxicidad que producen cada uno de estos agentes terapéuticos, tanto la relacionada con el «target» primario (dependiente del mecanismo de acción), como la independiente del mismo, permite entender las diferencias farmacodinámicas de cada uno de estos inhibidores. Esto permite mejorar el perfil de seguridad de estos fármacos e incluso combinarlos41.
¿Por qué es importante conocer la función tiroidea de pacientes oncológicos? Muchas veces el reconocimiento de hipotiroidismo solo por la clínica se vuelve dificultoso dado que los síntomas pueden estar causados no solo por el tumor en sí mismo, sino por el tratamiento antineoplásico o por la medicación utilizada para controlar síntomas acompañantes (náuseas o dolor) o, como en el caso de los ITC, por un efecto directo del fármaco. De forma similar, la clínica de tirotoxicosis se podría asemejar a ciertas complicaciones tumorales como sepsis o infecciones. Esto enfatiza que perder la posibilidad de realizar diagnóstico de disfunción tiroidea inducida por el tratamiento en estos pacientes llevaría injustamente a una reducción o la suspensión del fármaco utilizado como agente antineoplásico. Por otra parte, tener un hipotiroidismo o una tirotoxicosis no tratada podría afectar al metabolismo de otros fármacos reduciendo potencialmente su eficacia y además provocar consecuencias con riesgo de vida2.
Dada la alta prevalencia de disfunción tiroidea inducida por ITC sería recomendable realizar un perfil tiroideo antes de iniciar el tratamiento, durante el mismo y a las 2-4 semanas de finalizarlo, dado que en muchos pacientes la disfunción es solo transitoria, mientras que en algunos se torna definitiva42.
Luego del análisis de la bibliografía para la realización de esta monografía, de todos los mecanismos fisiopatológicos descriptos sobre cómo estos agentes orales producen hipotiroidismo, consideramos que el efecto antiangiogénico parecería ser el más sólido de los propuestos. Por tal motivo, el abordaje de estos pacientes debiera incluir no solo un examen físico y perfil tiroideo, sino también el estudio ecográfico de la glándula en forma periódica.
Por todo lo expuesto, sería necesaria la confección de guías de manejo de disfunción tiroidea inducida por ITC dirigidas tanto a endocrinólogos como a oncólogos que permitan un correcto abordaje de estos pacientes.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no poseer conflictos de intereses.