


























Introducción
Los tumores renales suponen la tercera causa de cáncer en la edad pediátrica, tras la leucemia y los tumores del sistema nervioso central. Entre los tumores renales primarios hasta un 90% aproximadamente son tumores de Wilms (TW), suponiendo el resto de tumores, no-Wilms, menos de un 10%1-3. Muchas de estas neoplasias renales primarias han sido clasificadas clásicamente como variantes del TW; sin embargo, en los últimos años se han ido reconociendo como entidades patológicas distintas. Estos tumores, aunque precisen de confirmación histológica para su diagnóstico, presentan unas características, tanto clínicas como epidemiológicas, y unos hallazgos radiológicos que permiten sugerir su diagnóstico en la mayoría de casos.
Material y métodos
Estudio retrospectivo de 26 pacientes con diagnóstico histológico de tumor renal distinto de Wilms recogidos en un hospital pediátrico de referencia durante los últimos 30 años. Se analizaron la forma de presentación, la edad, el sexo y los hallazgos radiológicos obtenidos mediante ecografía, tomografía computarizada (TC), resonancia magnética (RM) y urografía intravenosa (UIV). Los hallazgos evaluados fueron: tamaño, localización, vascularización, márgenes de la tumoración, presencia o no de necrosis, hemorragia, calcificaciones, hematoma subcapsular, adenopatías, metástasis a distancia y presencia de segundo tumor.
Todos los casos fueron confirmados con estudio histopatológico.
Resultados
En el período estudiado se diagnosticaron 150 tumores renales, de los cuales 124 fueron TW y 26 (17,3%) tumores distintos de Wilms.
De los 26 casos estudiados 11 fueron nefromas mesoblásticos, 7 correspondieron a tumor quístico multilocular, 4 a carcinoma de células renales, uno a sarcoma de células claras, otro a neuroblastoma intrarrenal, uno a tumor rabdoide con tumoración intracraneal sincrónica y otro a angiomiolipoma.
De los 11 nefromas mesoblásticos (con edad y presentación mostrados en la tabla 1) en 7 pacientes el tumor fue sólido y en 4 quístico, con presencia de un polo sólido. En 5 pacientes se encontraron áreas compatibles con necrosis, en dos sangrado intracavitario y en uno función excretora dentro de la masa. Siete pacientes tenían una colección subcapsular y 5 un anillo ecogénico periférico en la ecografía (figs. 1 y 2).
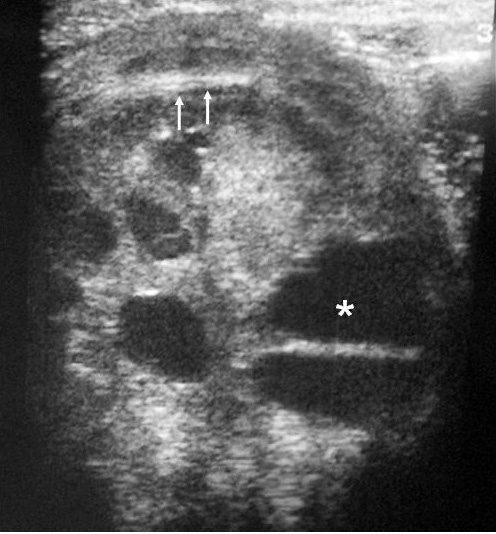
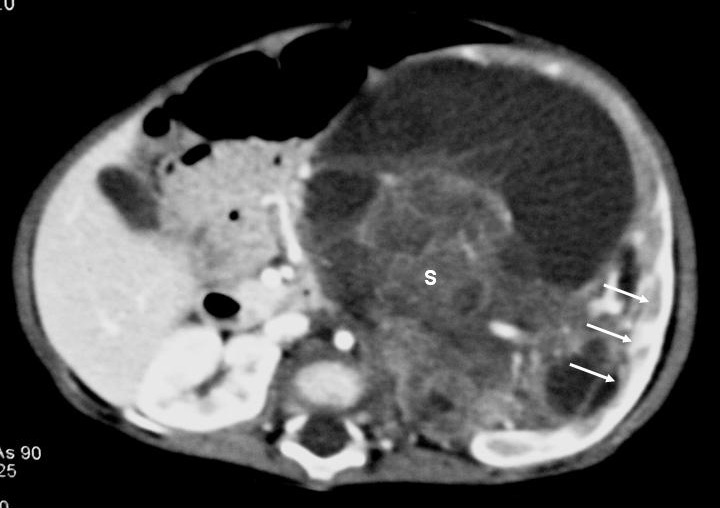
Fig. 1. Nefroma mesoblástico. Niño de 5 meses con masa abdominal palpable. (A) Ecografía abdominal, corte renal izquierdo. Masa en riñón izquierdo predominantemente sólida, con áreas de necrosis (*) y anillo ecogénico en la periferia (flechas). (B) Tomografía computarizada abdominal con contraste intravenoso. Corte al nivel del tercio medio del riñón. Masa en riñón izquierdo que cruza la línea media. Áreas de sangrado (S). Parénquima residual conservado en la periferia posterolateral de la masa (flechas).
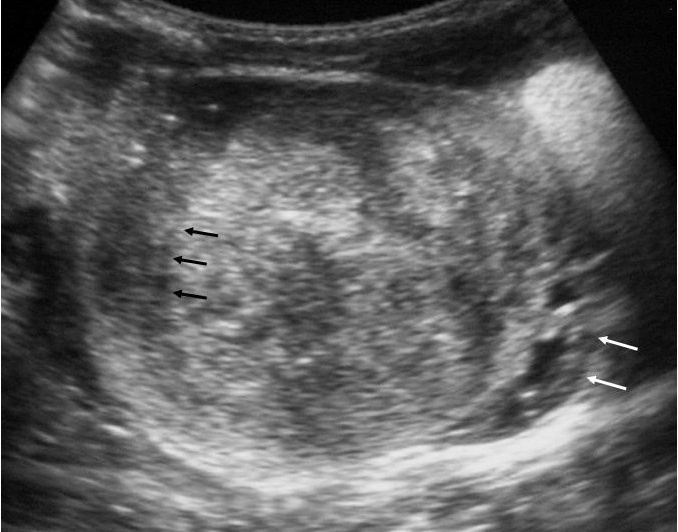
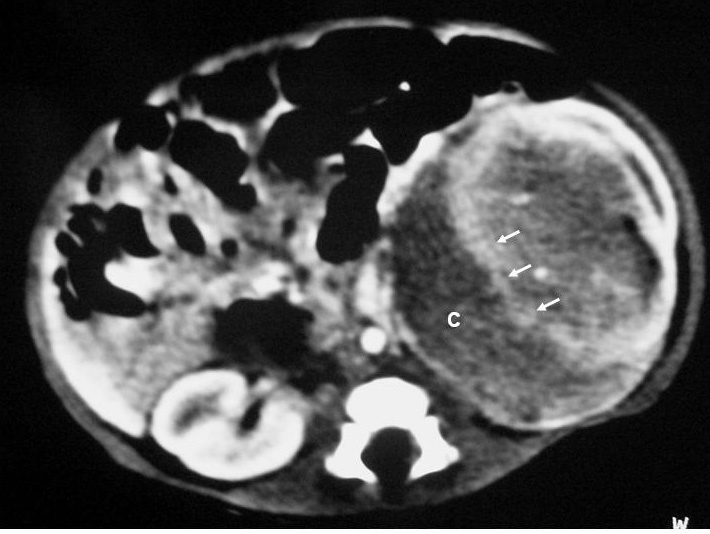
Fig. 2. Nefroma mesoblástico. Niña recién nacida con masa abdominal palpable, hipertensión arterial e hipercalcemia. (A) Ecografía abdominal. Corte renal longitudinal. Masa renal izquierda sólida, con colección subcapsular hipoecogénica (flechas negras). Resto de parénquima en polo inferior (flechas blancas). (B) Tomografía computarizada abdominal con contraste intravenoso. Corte al nivel del tercio medio renal. Masa renal hipodensa, con captación de contraste irregular, con anillo hipercaptante en la periferia del tumor (flechas blancas) y colección hipodensa subcapsular (C).

Se realizó nefrectomía a todos ellos. En un caso se precisó colectomía parcial, esplenectomía y suprarrenalectomía por infiltración tumoral. En dos casos se produjo rotura tumoral durante la intervención. Estos pacientes no recibieron tratamiento adyuvante.
Dos pacientes fallecieron por causas ajenas al tumor. En un paciente el estudio de la pieza quirúrgica demostró afectación de los bordes quirúrgicos, por lo que se trató con quimioterapia adyuvante. En el segundo control radiológico tras la cirugía se identificó recidiva tumoral local, por lo que fue nuevamente intervenido. Actualmente se le realizan controles radiológicos periódicos. El resto de pacientes permanecen sanos tras el tratamiento.
Los pacientes con diagnóstico histológico de tumor quístico multilocular (n = 7) tenían entre 5 meses y 12 años, siendo 4 de ellos varones y 3 mujeres. La clínica inicial fue la de masa abdominal palpable en 5, hematuria en uno y dolor abdominal en el paciente mayor. Se identificaron masas renales bien definidas que presentaban un aspecto quístico, con múltiples loculaciones separadas por septos que mostraban discreto realce (figs. 3 y 4). En un paciente se encontraron focos de blastema en las paredes de los septos en el estudio histológico.
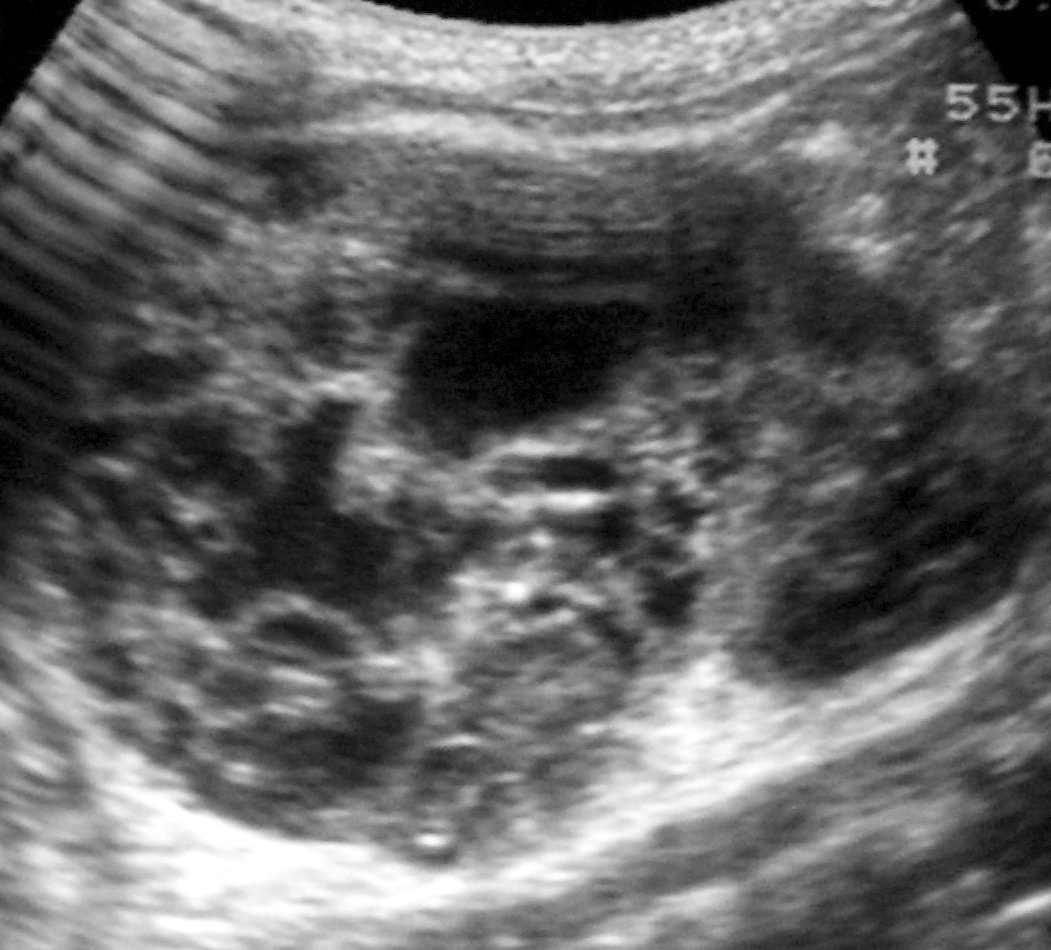
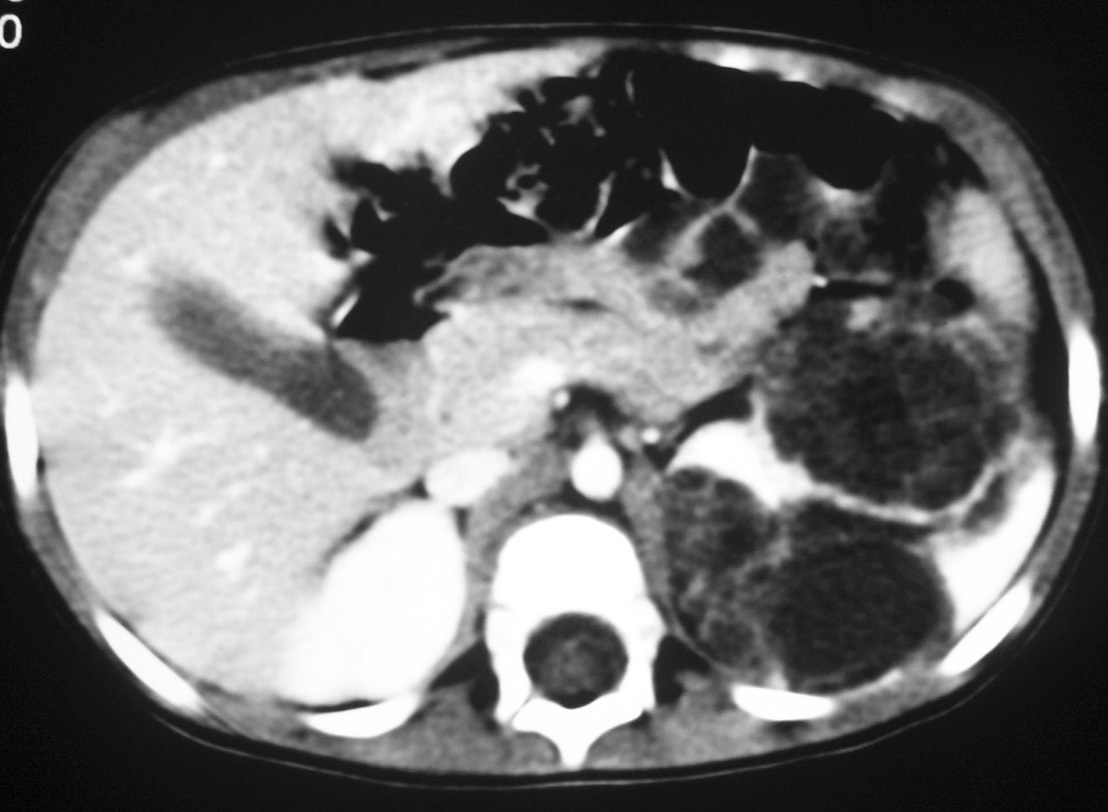
Fig. 3. Tumor quístico multilocular. (A) Ecografía abdominal. Corte renal longitudinal. Masa renal heterogénea con predominio de áreas anecoicas. (B) Tomografía computarizada abdominal con contraste intravenoso. Corte sobre polo superior renal izquierdo. Masa de aspecto multiquístico que presenta zonas de parénquima renal sano entre las formaciones quísticas.
Todos los pacientes quedaron libres de enfermedad tras la nefrectomía. En un caso pudo hacerse cirugía conservadora (tumorectomía) por tratarse de una masa de pequeño tamaño (fig. 4).
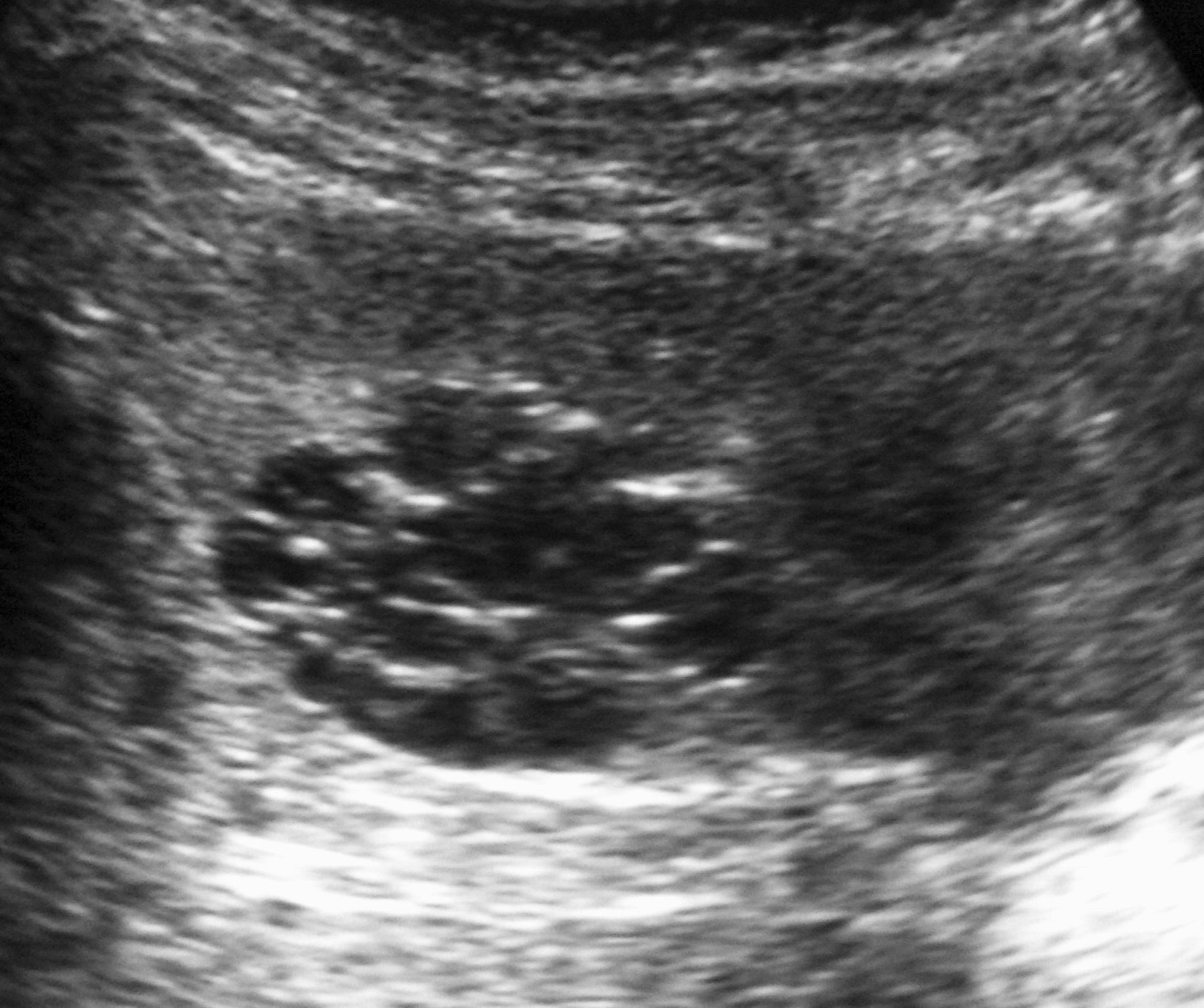
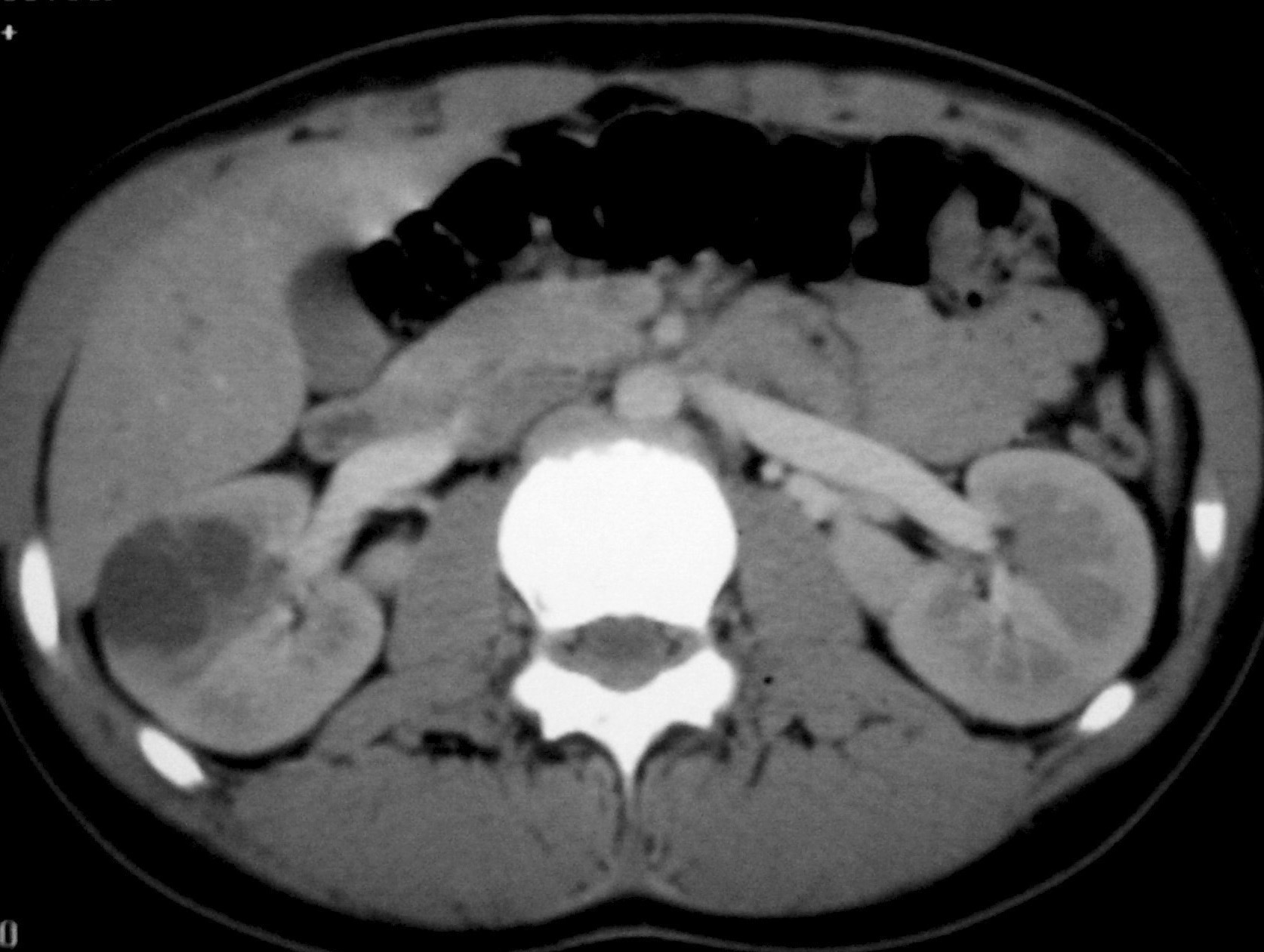
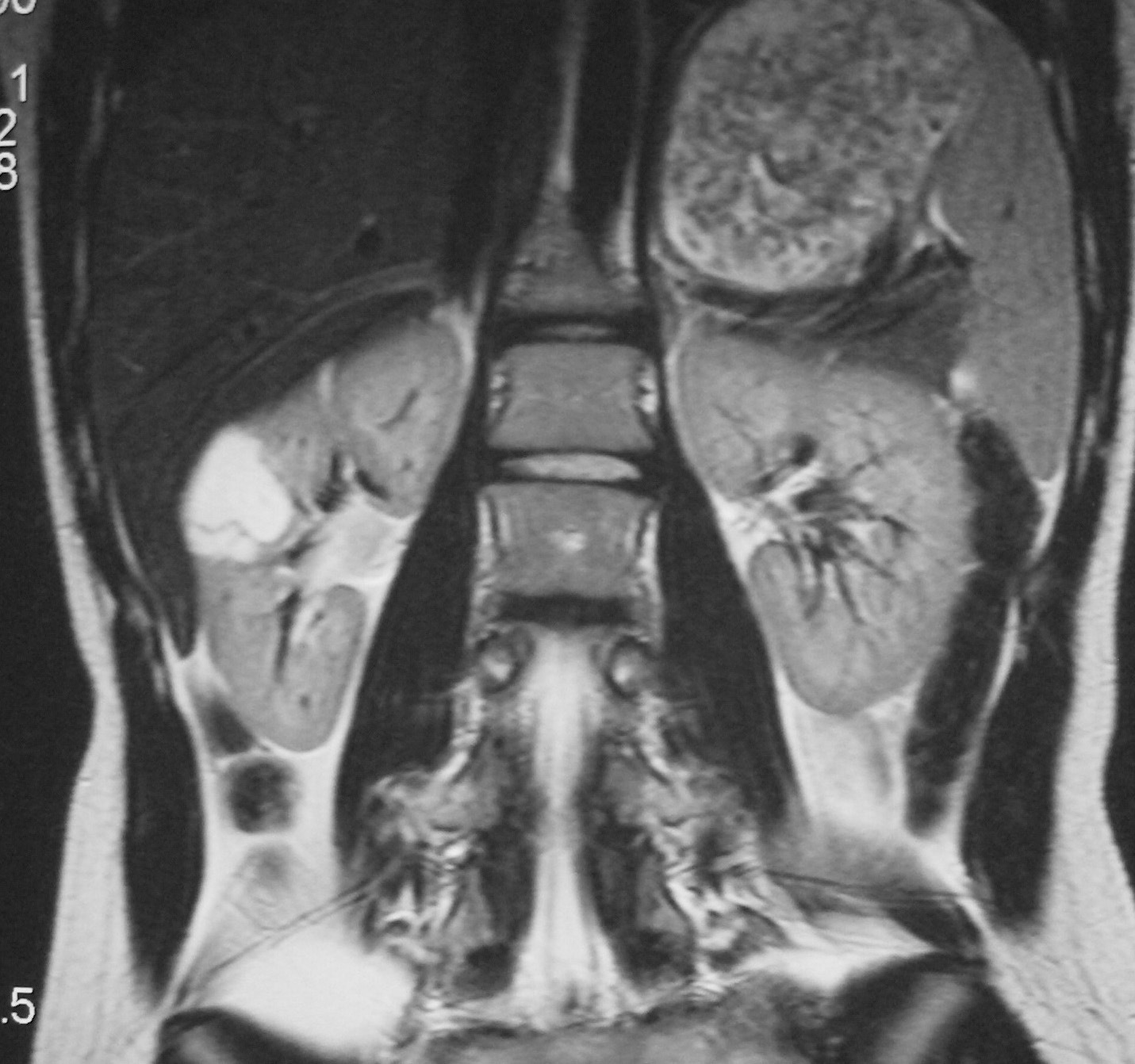
Fig. 4 Tumor quístico multilocular. (A) Ecografía abdominal. Corte renal longitudinal. Masa en tercio medio de riñón derecho, multiquística. (B) Tomografía computarizada abdominal con contraste intravenoso. Corte al nivel de los hilios renales. Cavidades hipodensas, tabicadas. (C) Resonancia magnética T2 coronal. Cavidades hiperintensas separadas por septos hipointensos bien definidos.
De los pacientes diagnosticados de carcinoma de células renales (n= 4) (con edad y presentación mostrados en la tabla 2) en dos casos se localizaron masas renales en la corteza renal, con infiltración del sistema pielocalicial. De estos dos, uno demostró fuga de contraste del sistema excretor en la radiografía abdominal post-TC con contraste, y el otro invasión de médula con extensión hasta el uréter proximal (fig. 5). En un único caso se apreciaron abundantes calcificaciones grumosas intratumorales dispersas (fig. 6).
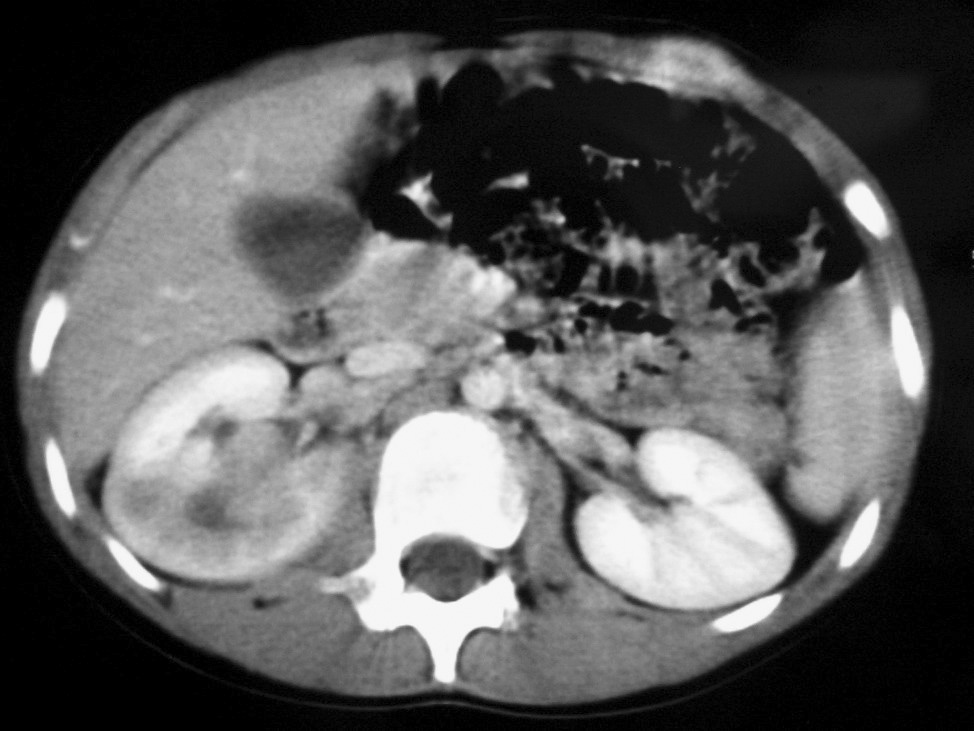
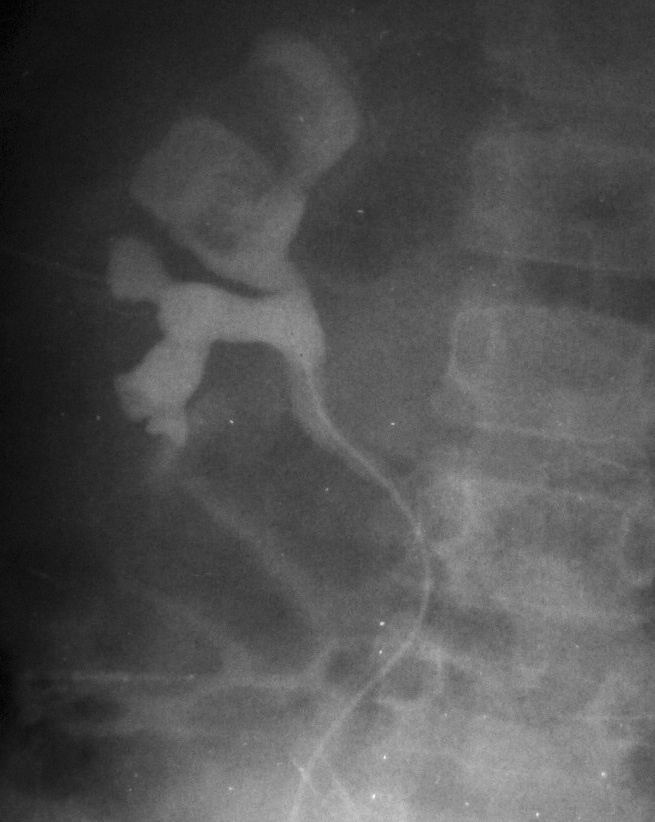
Fig. 5. Carcinoma de células renales. (A) Tomografía computarizada abdominal con contraste intravenoso. Corte axial sobre hilios renales. Riñón derecho aumentado de tamaño con discreto retraso de su función. Masa hipodensa central con extensión al sistema colector y escasa captación de contraste. (B) Pielografía ascendente del riñón derecho. En cálices del polo renal superior existe un defecto de repleción junto a dilatación y deformidad por tumoración que ocupa el sistema colector.
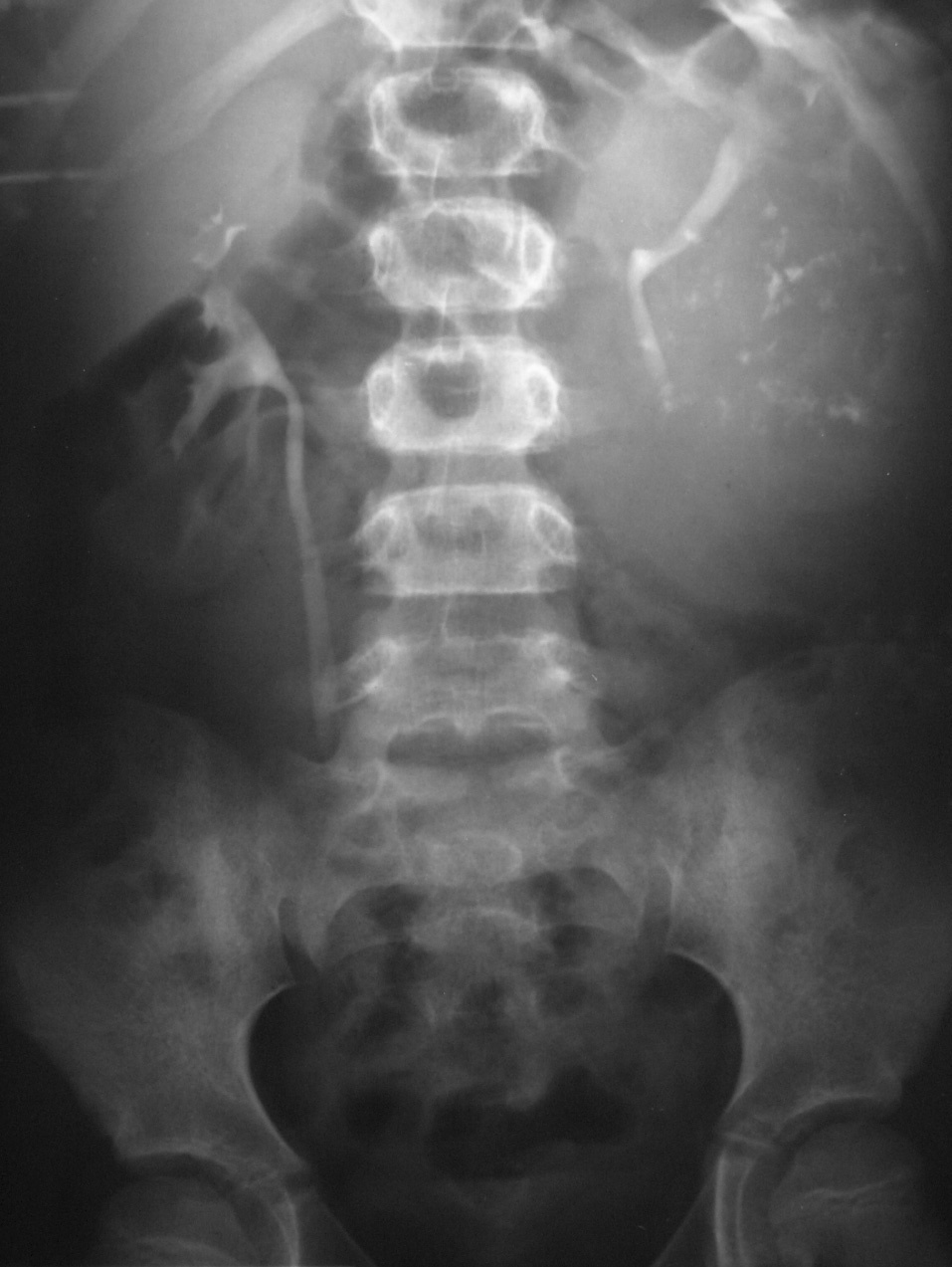
Fig. 6. Carcinoma de células renales. Urografía intravenosa que muestra una masa en hemiabdomen izquierdo, con abundantes calcificaciones grumosas dispersas, que desplaza el sistema pieloureteral.

Tras el tratamiento con nefrectomía y quimioterapia tres pacientes presentaron una remisión completa, y uno evolucionó de forma tórpida, falleciendo poco tiempo después del diagnóstico por diseminación general y multiorgánica.
El tumor rabdoide se presentó en un niño de 5 meses estudiado por masa abdominal palpable y hematuria, con el antecedente de un hermano gemelo fallecido poco tiempo antes por tumor cerebral. El estudio con ecografía y TC abdominal demostró una masa renal sólida, heterogénea e hipodensa con poco realce. Se completó el estudio con ecografía transfontanelar y TC craneal identificando una masa cerebral sincrónica, intraaxial, frontal paramedial, sólido-quística con calcificaciones (fig. 7). No se conoció la histología del tumor cerebral por seguimiento del paciente en otro centro.
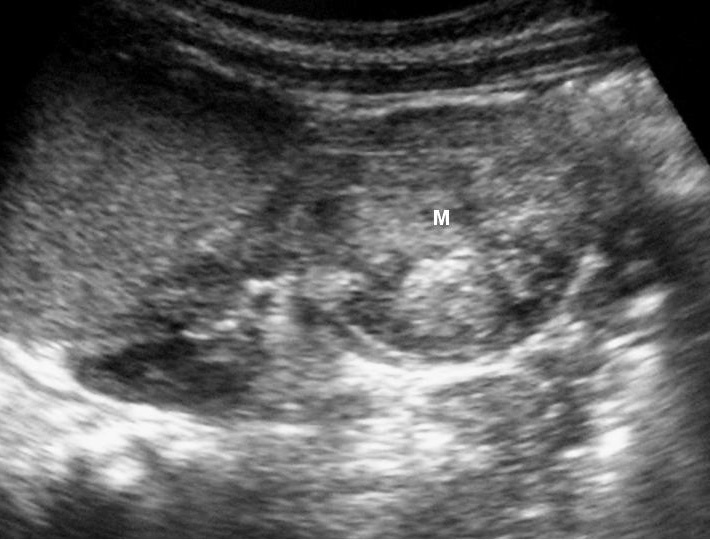
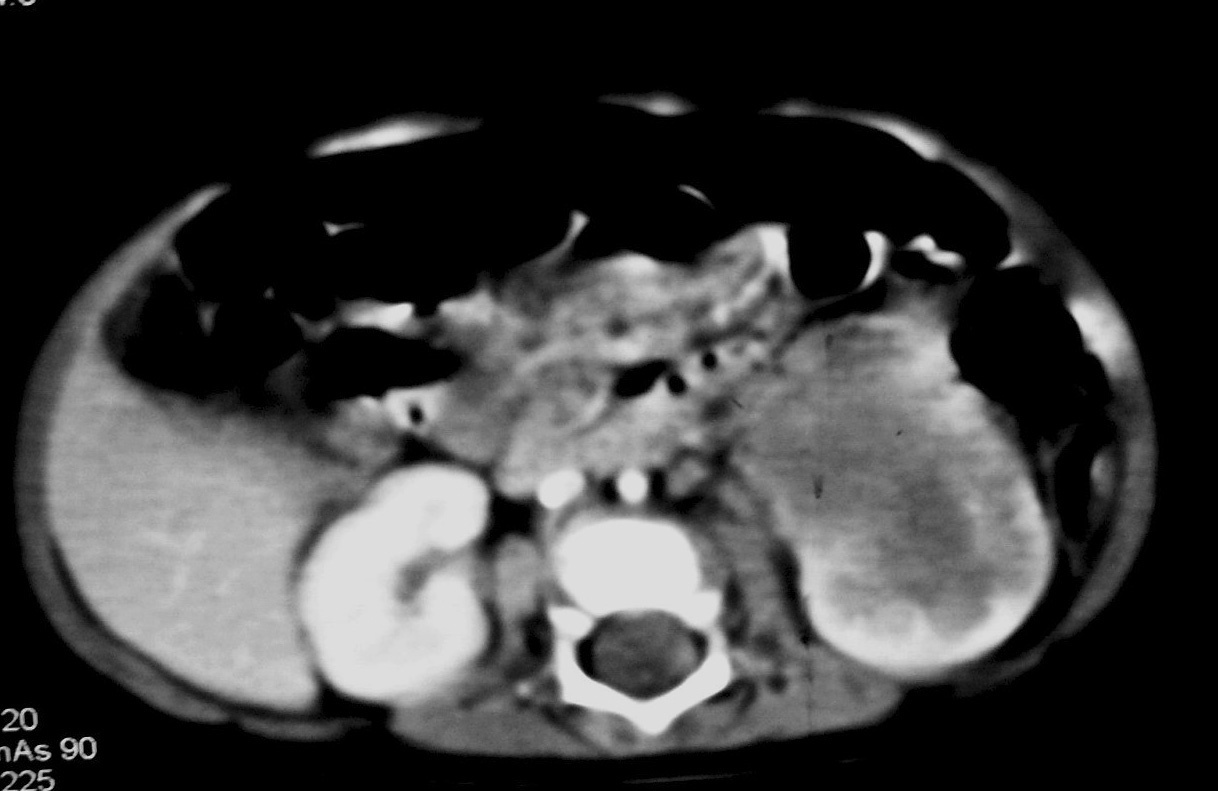
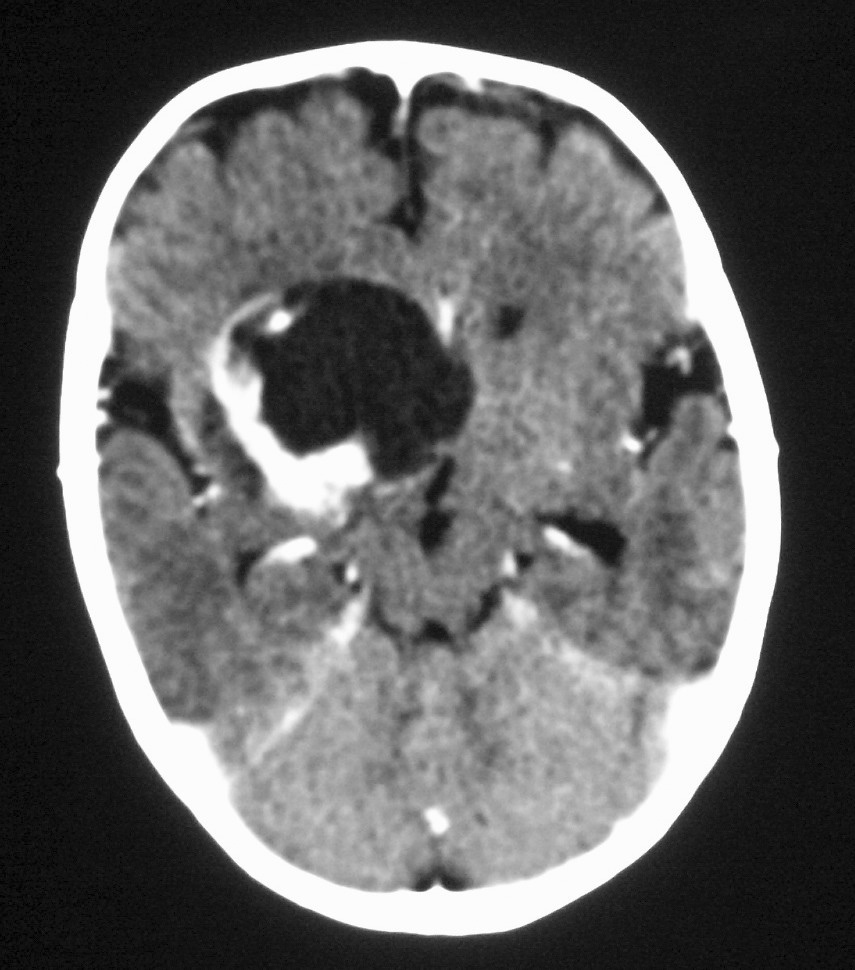
Fig. 7 Tumor rabdoide. Niño de 5 meses con hematuria y masa abdominal. (A) Ecografía abdominal. Corte longitudinal renal izquierdo. Masa sólida, heterogénea (M), bien delimitada en polo inferior de riñón izquierdo. (B) Tomografía computarizada abdominal con contraste intravenoso. Masa renal hipodensa, de aspecto infiltrativo, con crecimiento extrarrenal y tenue captación de contraste. (C) Tomografía computarizada craneal. Masa intraaxial, sólido-quística, fronto-temporal derecha, que desplaza la línea media, con un área de calcificación periférica.
El sarcoma de células claras tuvo lugar en un niño de 2 años estudiado por masa abdominal, en el que se identificó una masa intrarrenal, de predominio necrótico y una adenopatía retrocava (fig. 8).
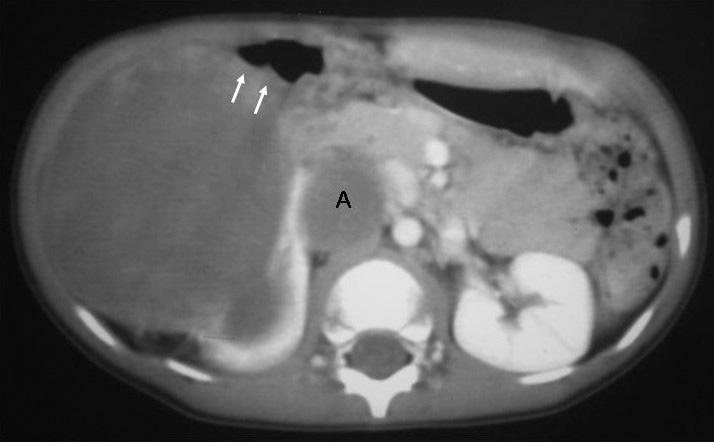
Fig. 8. Sarcoma de células claras. Tomografía computarizada abdominal con contraste intravenoso. Corte sobre polo inferior renal. Masa en riñón derecho, sólida, hipodensa, con gran crecimiento extrarrenal y que desplaza colon ascendente hacia delante (flechas). Adenopatía retrocava, hipodensa (A) de 3 cm.
La única paciente con angiomiolipoma renal fue una niña de 10 años con esclerosis tuberosa. En el riñón derecho se identificó una tumoración en el polo superior, con gran crecimiento extrarrenal, densidad homogénea, mayor que la del parénquima renal y menor que la del hígado y relativamente ecogénica (fig. 9). No se constataron áreas grasas, tratándose de un angiomiolipoma con poco componente graso. En el riñón izquierdo se identificaron dos lesiones de pequeño tamaño con atenuación grasa compatibles con angiomiolipomas. Como hallazgos asociados se observaron en el hígado múltiples lesiones con contenido graso de características similares, compatibles con angiomiolipomas intrahepáticos.
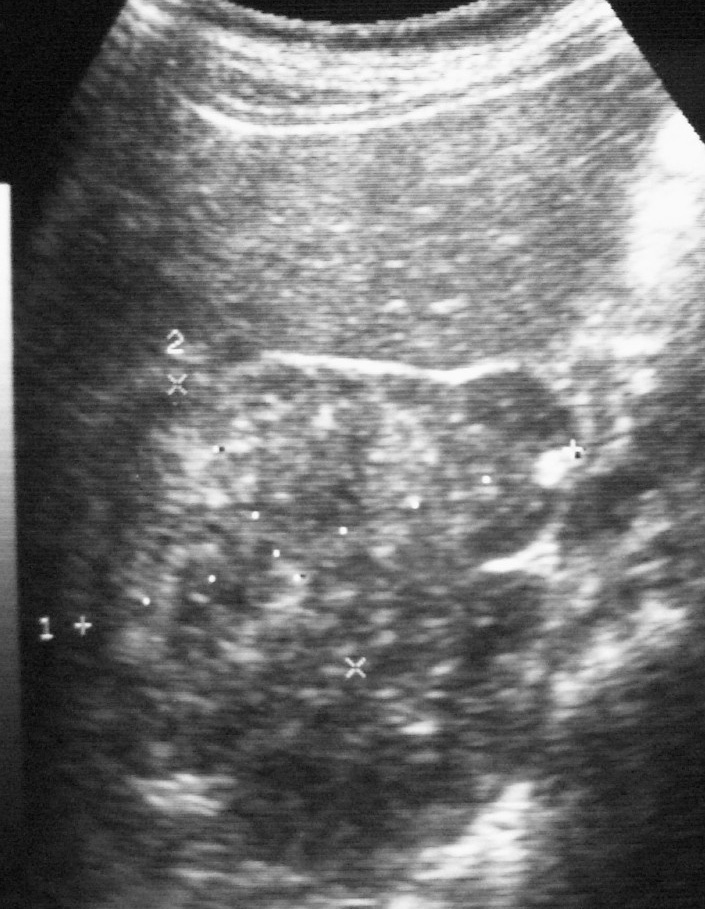
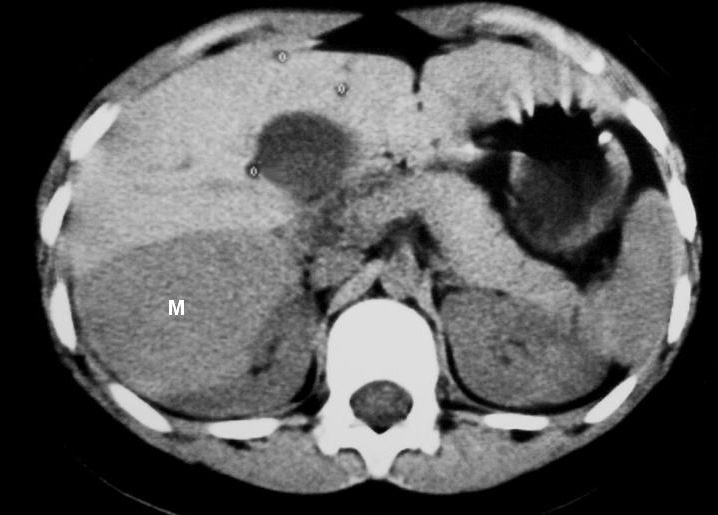
Fig. 9. Angiomiolipoma. Niña con esclerosis tuberosa. (A) Ecografía abdominal. Corte renal derecho. Masa renal de ecogenicidad heterogénea y contornos lobulados. (B) Tomografía computarizada abdominal. Masa (M) en riñón derecho. No se identifica componente graso. En el hígado existen tres lesiones de atenuación grasa correspondientes a angiomiolipomas hepáticos (1, 2, 3).
El paciente diagnosticado de neuroblastoma intrarrenal, un varón de 8 años de edad, acudió por fiebre y dolor en la pierna izquierda. Se identificó una masa en el riñón izquierdo, sólida, infiltrante, de ecogenicidad heterogénea, con áreas de necrosis y adenopatías hiliares (fig. 10). El estudio anatomopatológico de un cilindro obtenido mediante biopsia renal no pudo diferenciar entre TW y neuroblastoma, por lo que se realizó estudio isotópico con meta-yodo-bencil-guanidina (MIBG) que resultó positivo, con infiltración de la médula ósea. Después de varios ciclos de quimioterapia la masa disminuyó de tamaño y se identificó la presencia de algunas calcificaciones.
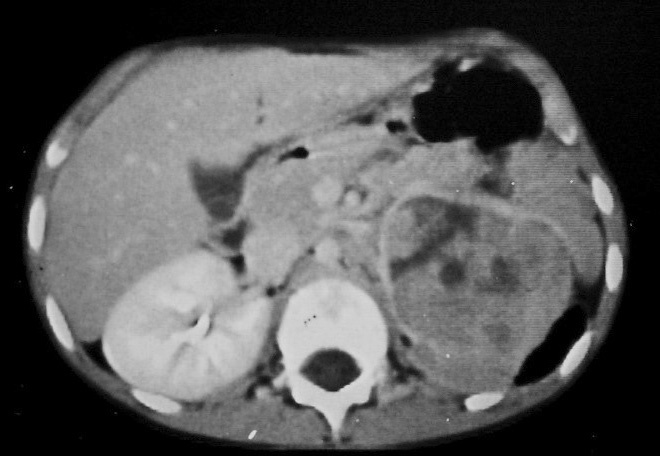
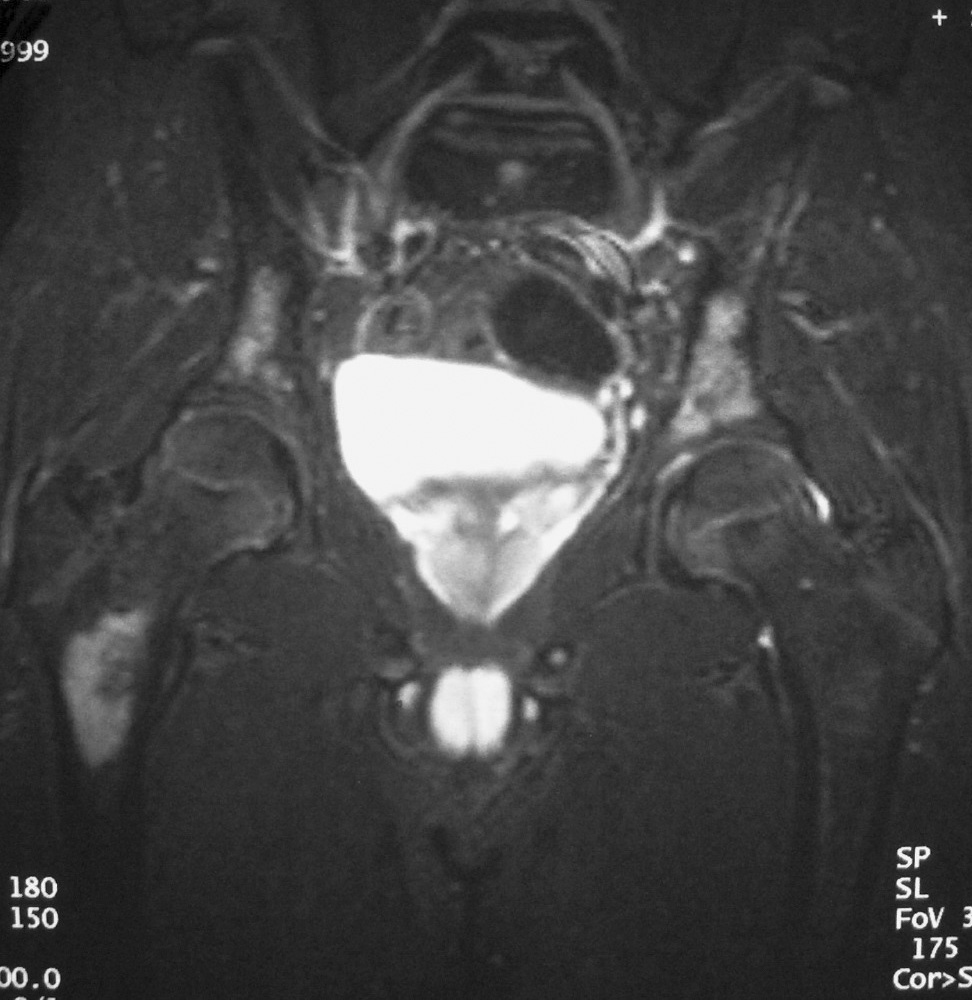
Fig. 10. Neuroblastoma intrarrenal. Niño de 9 años que presenta fiebre y dolor en la pierna izquierda. (A) Tomografía computarizada abdominal con contraste intravenoso. Masa intrarrenal sólida, con áreas necróticas, con mínimo parénquima circundante. (B) Resonancia magnética de control tras tratamiento con quimioterapia. Secuencia STIR. Cortes coronales de pelvis y ambas caderas. Lesiones hiperintensas en la medula ósea próxima a trocánter menor derecho, parte superior de hueso iliaco izquierdo y ambas regiones acetabulares.
Tras una evolución tórpida y con desarrollo de enfermedad metastásica múltiple, falleció 8 años después del diagnóstico.
Discusión
A continuación se revisan los aspectos clínicos y epidemiológicos y los hallazgos radiológicos más importantes de los tumores renales distintos de Wilms, que pueden ayudar al radiólogo en su reconocimiento.
Nefroma mesoblástico
Es el tumor sólido más frecuente del neonato. Habitualmente es diagnosticado en los tres primeros meses de vida, siendo hasta el 90% de los casos diagnosticados dentro del primer año1,4. Todos los pacientes diagnosticados en nuestro centro tenían menos de 1 año, y 10 de ellos (90,9%) menos de 3 meses. Clínicamente suele presentarse como una masa abdominal palpable, que puede acompañarse de hematuria, hipercalcemia e hipertensión en algunas ocasiones.
Embriológicamente es un tumor con origen en el mesénquima nefrogénico. En ocasiones, el tumor puede englobar el parénquima renal, el cual mantendrá su capacidad de excreción activa. Macroscópicamente es sólido, infiltrativo, con márgenes mal definidos y no encapsulado. Existen dos variantes: clásica y celular. La apariencia de la variante clásica recuerda a un leiomioma, mientras que la celular presenta mayor número de mitosis, células apoptóticas y necrosis, siendo potencialmente más agresiva1,5,6.
Los estudios de imagen muestran una masa por lo general de gran tamaño, sólida, que suele englobar el seno renal y puede contener, aunque no es frecuente, áreas quísticas, hemorrágicas y necróticas. No presenta buena delimitación con el parénquima sano y puede existir infiltración local de tejidos vecinos1. En uno de nuestros pacientes, estudiado por masa abdominal y cuadro clínico de obstrucción intestinal, se identificó una masa renal que infiltraba el colon, el bazo y la glándula suprarrenal. Un signo ecográfico característico es la imagen en anillos concéntricos hiper e hipoecoicos en la periferia del tumor5,7. En nuestra serie la ecografía demostró una colección hipoecoica subcapsular en 7 pacientes y anillo ecogénico periférico en 5.
Tanto la recurrencia local como las metástasis, descritas en pulmón, cerebro y hueso, son raras. Sólo en un caso se produjo una rediciva local. En ningún caso se hallaron metástasis a distancia.
El tratamiento de elección es la nefrectomía con márgenes amplios. En los casos de variante celular, o aquellos en que se muestren los bordes quirúrgicos afectos, se recomienda tratamiento con quimioterapia adyuvante3. Se aconseja seguimiento postquirúrgico frecuente al menos durante el primer año en todos los casos1.
El pronóstico es muy bueno, obteniendo excelentes resultados tras la nefrectomía con márgenes amplios.
Tumor quístico multilocular
Tumor benigno que se presenta con dos picos de incidencia, uno de los 3 meses a los 4 años, de predominio en varones, y otro en adultos, a los 40 años, de predominio en mujeres4. En nuestro grupo de pacientes la edad media al diagnóstico fue de 2 años y 9 meses.
Clínicamente se presenta como una masa unilateral y solitaria que no se acompaña de dolor ni síndrome constitucional. Puede existir hematuria y otros síntomas por obstrucción del sistema colector renal.
El estudio anatomopatológico muestra una masa encapsulada, quística, multiloculada, formada por quistes no comunicados que están tapizados por epitelio plano cuboide y separados por septos. Cuando los septos se componen sólo de tejido fibroso se trata de un nefroma quístico, mientras que si existen focos de blastema en las paredes de los quistes es un nefroblastoma quístico parcialmente diferenciado1,6. Se ha asociado un mayor riesgo de comportamiento agresivo del nefroma quístico parcialmente diferenciado con la cantidad de tejido inmaduro que contenga8.
Los estudios radiológicos muestran una masa de gran tamaño, encapsulada, con múltiples quistes no comunicantes, que pueden herniarse hacia la pelvis y el uréter y cuyos septos presentan un realce irregular bajo o moderado4,8,9.
Los hallazgos encontrados en los estudios de imagen no permiten la distinción entre nefroma quístico y nefroblastoma quístico parcialmente diferenciado, aunque esto no tiene relevancia de cara al tratamiento, puesto que es el mismo: la excisión quirúrgica6,8,9. Sin embargo, el nefroma quístico parcialmente diferenciado puedemuy bueno tras la nefrectomía.
Sólo en uno de nuestros pacientes se encontraron focos de blastema en el estudio histológico. A un paciente se le realizó tumorectomía y el resto fueron tratados con nefrectomía. Todos ellos quedaron libres de enfermedad tras la cirugía.
Es muy importante realizar el diagnóstico diferencial con otras masas renales que requieren un tratamiento más agresivo, como el TW quístico, el sarcoma de células claras o el carcinoma de células renales quístico8-10.
Carcinoma de células renales
Es un tumor infrecuente en la edad pediátrica, con una relación con el TW de 30:1. En la segunda década de vida esta relación se iguala, por lo que es muy importante considerarlo en el diagnóstico diferencial de tumores renales en niños mayores. La mayoría son esporádicos, aunque se han descrito asociaciones con el síndrome de von Hippel-Lindau y la esclerosis tuberosa1,11.
Las manifestaciones clínicas típicas son dolor en flanco, masa abdominal y hematuria, siendo esta última más frecuente que en el TW.
Se cree que se origina de las células epiteliales de los túbulos renales y se reconocen distintos subtipos celulares6,12.
Suele ser más pequeño que el TW al diagnóstico11. En los estudios radiológicos se suele presentar como una masa sólida, infiltrante, con áreas de necrosis, hemorragia, degeneración quística y calcificaciones, que tienden a ser más densas y homogéneas que en el TW e incluso pueden verse en ganglios con afectación metastásica13. Invade localmente con extensión al retroperitoneo y los ganglios linfáticos. Metastatiza de forma más frecuente en los pulmones, el hueso, el hígado y el cerebro1.
En dos de nuestros pacientes, que presentaron hematuria franca, hubo extensión tumoral hacia los cálices y la pelvis renal, así como afectación ganglionar del retroperitoneo. Los dos casos se trataron con nefrectomía con buena evolución.
Los pacientes que tienen un tumor localizado en el riñón tienen buen pronóstico comparado con aquellos que tienen afectación de los ganglios linfáticos regionales o metástasis a distancia12, puesto que la nefrectomía con linfadenectomía regional ofrece buenos resultados y el tumor es muy resistente a la quimioterapia1.
Tumor rabdoide
Supone el 2-3% de todas las masas renales en la edad pediátrica y es el tumor renal de peor pronóstico1,4. Aproximadamente el 80% tiene lugar en menores de 2 años y el 60% en menores de 1 año, siendo muy infrecuente en mayores de 5 años14.
Puede presentarse con hematuria o hipercalcemia paraneoplásica, por elevación de niveles de parathormona, pero debido a su agresividad es frecuente que se muestre con los síntomas derivados de las metástasis. Se asocia a la aparición sincrónica o metacrónica de un tumor cerebral primario, generalmente en la línea media y frecuentemente en la fosa posterior, que puede tratarse de un tumor neuroectodérmico primitivo, ependimoma, astrocitoma del troncoencéfalo o cerebeloso o meduloblastoma4,14,15.
El paciente que presentamos se estudió por hematuria y masa abdominal palpable a los 5 meses de edad. En el momento del diagnóstico se encontró una masa renal y una masa cerebral en el lóbulo frontal.
Histológicamente está compuesto por células que recuerdan a las de los tumores musculoesqueléticos. Macroscópicamente es una masa que suele crecer del parénquima perihiliar, infiltrando la médula, el seno renal y el sistema colector. Al diagnóstico suele ser grande e invadir todo el parénquima renal14. Los estudios de imagen muestran una gran masa lobulada, heterogénea, con áreas de necrosis y hemorragia y, en ocasiones, con calcificaciones lineales. Es característica, aunque no patognomónica, la existencia de una colección líquida subcapsular periférica, que puede representar un hematoma o bien restos tumorales necrosados14-16. La existencia de esta colección, junto a la hipercalcemia y el rango de edad, son hallazgos característicos que comparte este tumor con el nefroma mesoblástico. Sin embargo, dada la baja frecuencia de estos tumores, ante una colección líquida periférica en un niño pequeño con masa renal, el diagnóstico diferencial más probable será el de nefroma mesoblástico5,15. En nuestra serie de los 11 nefromas mesoblásticos encontrados 7 (63,3%) tenían una colección líquida subcapsular, mientras que el único caso de tumor rabdoide se presentó como una masa intrarrenal sin colección periférica.
Las metástasis del tumor rabdoide son frecuentes y tempranas, y los órganos más afectados son el pulmón, el hígado, el cerebro, el hueso y los ganglios linfáticos1.
El pronóstico es muy malo, teniendo una supervivencia del 20% a los 18 meses4.
Sarcoma de células claras
Es un tumor muy agresivo que supone un 4% de todos los tumores renales en niños, con mayor incidencia entre 1 y 4 años y con predominio en varones1. Se conoce también como tumor metastatizante óseo de la infancia, por su gran avidez para metastatizar en el hueso.
Macroscópicamente es indistinguible del TW unilateral y único1,2,10. Es un tumor sólido, aunque hasta en un 50% puede tener quistes, no encapsulado, que infiltra, distorsiona y comprime el parénquima renal4. Puede tener áreas de necrosis y hemorragia y no suele haber invasión intravascular1,4.
Su comportamiento es muy agresivo y su pronóstico es peor que el del TW. Aunque no suele existir al diagnóstico, es muy característica la afectación metastásica del hueso, ya sea con un patrón lítico, permeativo o escleroso1,10. También puede metastatizar en los ganglios, el hígado y el pulmón. Requiere un largo seguimiento tras el tratamiento quirúrgico y quimioterápico, puesto que las metástasis pueden presentarse mucho tiempo después del tratamiento.
En el paciente de nuestra serie, con una masa renal grande, de aspecto necrótico y una gran adenopatía retroperitoneal, no se encontraron lesiones óseas asociadas. Se trató con cirugía, radioterapia y quimioterapia con buena evolución.
Angiomiolipoma
Es una lesión hamartomatosa rara en la edad pediátrica de forma esporádica, pero presente en un 40-80% de pacientes con esclerosis tuberosa y también asociada a neurofibromatosis y enfermedad de von Hippel-Lindau1,2,6. En los pacientes que asocian estos síndromes existe un pico de incidencia a los 10 años1.
Cuando se presenta de forma esporádica suele ser una masa renal única, mientras que los pacientes con esclerosis tuberosa suelen tener una afectación múltiple y bilateral2.
Se compone de distinta cantidad de tejido adiposo, vasos y músculo liso. En ocasiones los vasos aberrantes de su interior dan lugar a formaciones aneurismáticas. El 95% de estos tumores tiene focos grasos, distinguibles con ecografía, TC y RM. En aquellos casos en que la TC sin contraste muestre zonas grasas con unos valores de atenuación menores a 20 unidades Hounsfield se podrá hacer un diagnóstico específico10. En caso de que la lesión no tenga grasa o la tenga en cantidades indetectables macroscópicamente, como en el caso de nuestra serie, los hallazgos serán más inespecíficos, pudiéndose confundir con una masa sólida. Es importante tener en cuenta que existen TW que tienen zonas grasas, por lo que ante una sospecha de angiomiolipoma que presenta un comportamiento atípico debe plantearse el diagnóstico diferencial con el TW2.
El tratamiento, nefrectomía o embolización sólo es preciso en aquellas lesiones con riesgo de sangrado, por lo que es aconsejable el seguimiento ecográfico cada dos o tres años en los pacientes con esclerosis tuberosa, y anual tras la pubertad para detectar posibles crecimientos.
Neuroblastoma intrarrenal
Es una tumoración renal muy infrecuente cuyo origen se cree que tiene lugar a partir de restos de tejido adrenal que quedan atrapados en el tejido renal, o de ganglios simpáticos intrarrenales17.
Se presenta con masa abdominal palpable, que puede asociar hipertensión y elevación de catecolaminas, de ácido vanilmandélico y homovanílico en sangre y orina.
Los neuroblastomas tienen lugar en un 50-70% en las glándulas suprarrenales e histológicamente pertenecen al grupo de los tumores de células pequeñas, redondas y azules de la infancia (como sarcoma de Ewing, rabdomiosarcomas y linfomas).
El neuroblastoma intrarrenal se muestra en los estudios de imagen como una masa renal que puede presentar calcificaciones y que puede ser indistinguible del TW en los casos histológicamente más indiferenciados y, por lo tanto, de peor pronóstico18. En el paciente de nuestra serie se encontró una masa renal sólida e infiltración de médula ósea. En el estudio de la pieza quirúrgica se comprobó que la glándula suprarrenal no estaba afectada.
Estos tumores metastatizan y el pronóstico es muy malo.
Conclusión
Pese a la baja incidencia que presentan otros tumores renales en la edad pediátrica frente al TW es importante su detección de cara a establecer el tratamiento, de tipo conservador o quirúrgico, y el pronóstico de los mismos. El diagnóstico diferencial de las lesiones focales renales debe incluir tanto procesos tumorales como no tumorales, como pueden ser procesos inflamatorios localizados o quistes.
Los hallazgos radiológicos, junto a datos clínicos y epidemiológicos, tienen un papel importante para orientar el diagnóstico de estas masas.
El diagnóstico histológico sigue siendo indispensable.

