








Definición
Los analgésicos son principios activos que disminuyen o inhiben la percepción del dolor. Con el tratamiento se persigue la anulación específica de la percepción del dolor, pero sin afectar a otras capacidades sensitivas como la percepción de los estímulos táctiles o de los estímulos térmicos. El término dolor es el punto débil de la definición. Lo utilizamos para describir un amplio abanico de sensaciones. Hablamos de dolor tanto después de recibir una patada en la espinilla como después de que nos comuniquen la noticia del fallecimiento de un ser querido. La definición del término como la publicada por la Sociedad Internacional para el Estudio del Dolor intenta precisar el concepto en el sentido de describir el dolor como una sensación desagradable que se asocia a un daño tisular o a un daño de este tipo.
Generación, conducción y percepción del dolor
De acuerdo con esta definición, se genera dolor en el momento en que las percepciones sensitivas fisiológicas, es decir, los estímulos mecánicos, térmicos, químicos o eléctricos, superan un determinado valor umbral. Esto se acompaña a menudo de un daño tisular y lleva sistemáticamente a la liberación de sustancias que, como mediadores del dolor, pueden potenciar o mantener la percepción del dolor.
Se percibe una sensación de dolor si una célula o un grupo pequeño de células generan un potencial de acción en respuesta a un estímulo y transmiten la excitación eléctrica a las neuronas aferentes. Estas estructuras periféricas se denominan receptores. En la fisiología se utiliza el término receptor para definir una célula o un grupo de células capaz de generar un potencial de acción en respuesta a un estímulo. En términos fisiológicos, el receptor es un elemento histológico. En cambio en la farmacología, se designa como receptor al interlocutor molecular que reacciona con sustancias biológicamente activas. Estos receptores pueden estar especializados en la percepción de determinados estímulos (receptores del frío, propiorreceptores) o pueden percibir múltiples cambios que se producen en el entorno (receptores polimodales) y convertirlos en un estímulo eléctrico (potencial generador).
Los receptores especializados en la percepción del dolor (nociceptores) se localizan en el extremo periférico de las neuronas aferentes y su activación da lugar a potenciales de acción que se transmiten al sistema nervioso central después de hacer sinapsis en los ganglios del asta posterior (fig. 1). Las neuronas aferentes forman parte del grupo de las fibras C y las fibras Aq delgadas, no mielinizadas. Como ya se ha comentado, la excitación de los nociceptores puede provocar la liberación de mediadores del dolor endógenos (histamina, H+, K+, prostaglandinas, bradicinina y otros muchos) que a su vez potencian la señal primaria a través de la disminución del umbral de excitación o al aumentar la eficiencia de la transmisión sináptica de la excitación. Los mecanismos indicados rigen para la percepción de dolores «fisiológicos», es decir, de dolores que se perciben cuando los estímulos fisiológicos superan el umbral de dolor. Este dolor se puede calificar de fisiológico por representar una función de protección y alerta natural en el ser humano.
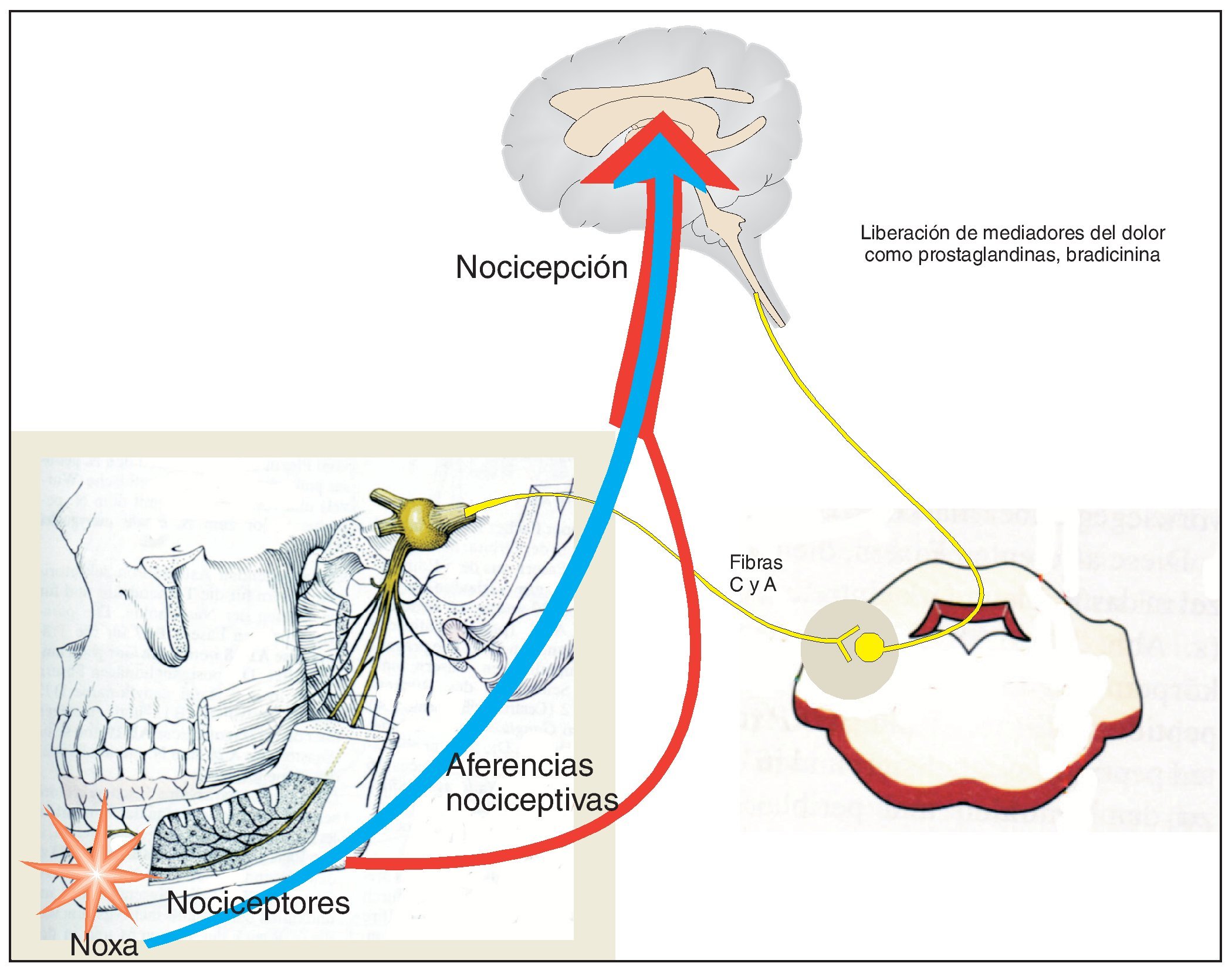
Figura 1. Representación esquemática de las estructuras que intervienen en la nocicepción. Diversos tipos de noxas activan los nociceptores, cuyas señales eléctricas son enviadas a la corteza cerebral a través de neuronas sensitivas tras varias sinapsis. El esquema simplifica considerablemente los hechos reales. Por ejemplo, las neuronas sensibles que intervienen en la nocicepción no constituyen una población uniforme. Difieren por la dotación de proteínas funcionales integradas en su membrana y también se diferencian por las zonas del cerebro a las que envían la información. Determinadas neuronas sensibles transmiten información sobre la localización y la intensidad del estímulo doloroso, mientras que otras están relacionadas con la componente afectiva, es decir, el potencial de amenaza del dolor percibido. La noxa no desencadena sólo una actividad eléctrica en forma de potenciales de acción, sino que también libera mediadores del dolor que pueden sensibilizar el sistema nociceptivo y contribuir a aumentar la percepción del dolor. De estos mediadores forman parte productos de la enzima ciclooxigenasa como las prostaglandinas. La inhibición de la ciclooxigenasa es un mecanismo esencial del efecto analgésico. Los analgésicos actúan también a nivel de las sinapsis del sistema nociceptivo. Determinados analgésicos (opioides) atenúan la unión sináptica.
Nociceptores y dolor neuropático
El dolor adquiere carácter patológico si los mediadores liberados por el estímulo primario modifican de tal manera el sistema nociceptivo que se perciben como dolorosos estímulos que normalmente no superan el umbral crítico (hiperalgesia, alodinia) y el acontecimiento doloroso se realimenta (cronificación). La inducción de las ciclooxigenasas es uno de los mecanismos participantes en la sensibilización y la cronificación. Estas enzimas forman a partir de un componente celular omnipresente, el ácido araquidónico, una serie de productos denominados prostaglandinas que incrementan la excitabilidad de los nociceptores entre otros efectos. Este tipo de dolor es un dolor nociceptivo patológico.
Las alteraciones del sistema nociceptivo también pueden provocar dolor. Dichas alteraciones activan procesos de reparación a través de la liberación de factores de crecimiento y citocinas. Se modifica el patrón de proteínas funcionales en las neuronas aferentes, por ejemplo, el recubrimiento con canales de iones y receptores o la actividad enzimática. Se ha podido demostrar que después de la contusión de neuronas aferentes se expresa en las neuronas funcionales residuales o en las neuronas neoformadas un tipo distinto de canal de sodio. Este canal de sodio podría ser corresponsable de la hiperalgesia patológica en el ser humano después de un traumatismo neuronal como resultado de cambios en el comportamiento regulador. Si los distintos procesos, indispensables para una reparación eficaz, se ejecutan de forma descoordinada, la restitución no es posible y el estado de hipersensibilidad patológica se mantiene. Esta forma de dolor se denomina dolor neuropático.
Es evidente que el enfoque terapéutico del dolor nociceptivo patológico y del dolor neuropático no puede ser igual. Sin embargo, se ha de advertir que el dolor nociceptivo puede tener un componente neuropático y viceversa, el dolor neuropático también puede acompañarse de dolores nociceptivos. Sin embargo, la separación de ambas formas de dolor parece lógica, dado que exigen enfoques terapéuticos totalmente diferentes.
En la odontología es relativamente fácil identificar el origen de un dolor (nociceptivo), que puede ser erradicado mediante un tratamiento odontológico. Se debe dar preferencia siempre al tratamiento etiológico frente al tratamiento farmacológico sintomático. La prescripción de un analgésico debe constituir sólo una medida complementaria.
Dolor nociceptivo
Para el tratamiento del dolor nociceptivo se dispone de seis principios activos farmacológicos distintos que se comentan más detalladamente a continuación.
Inhibidores de la ciclooxigenasa
El incremento de la síntesis de mediadores del dolor del tipo de las prostaglandinas por inducción de la enzima ciclooxigenasa es un factor que contribuye a la aparición del dolor nociceptivo patológico, por lo que es razonable esperar que los inhibidores de la ciclooxigenasa mejoren este tipo de dolor. Esto es válido para todo el grupo de los inhibidores de la ciclooxigenasa, empezando por el ácido acetilsalicílico introducido en el tratamiento analgésico hace más de 100 años.
El problema de este grupo de sustancias es que las prostaglandinas no sólo tienen efectos patológicos, en este caso potenciadores del dolor, sino también funciones fisiológicas importantes como la protección de la mucosa gástrica, la perfusión renal y la dilatación de las vías respiratorias (fig. 2). La afectación de los efectos fisiológicos explica el patrón de efectos adversos de los inhibidores de la ciclooxigenasa y de sus contraindicaciones:
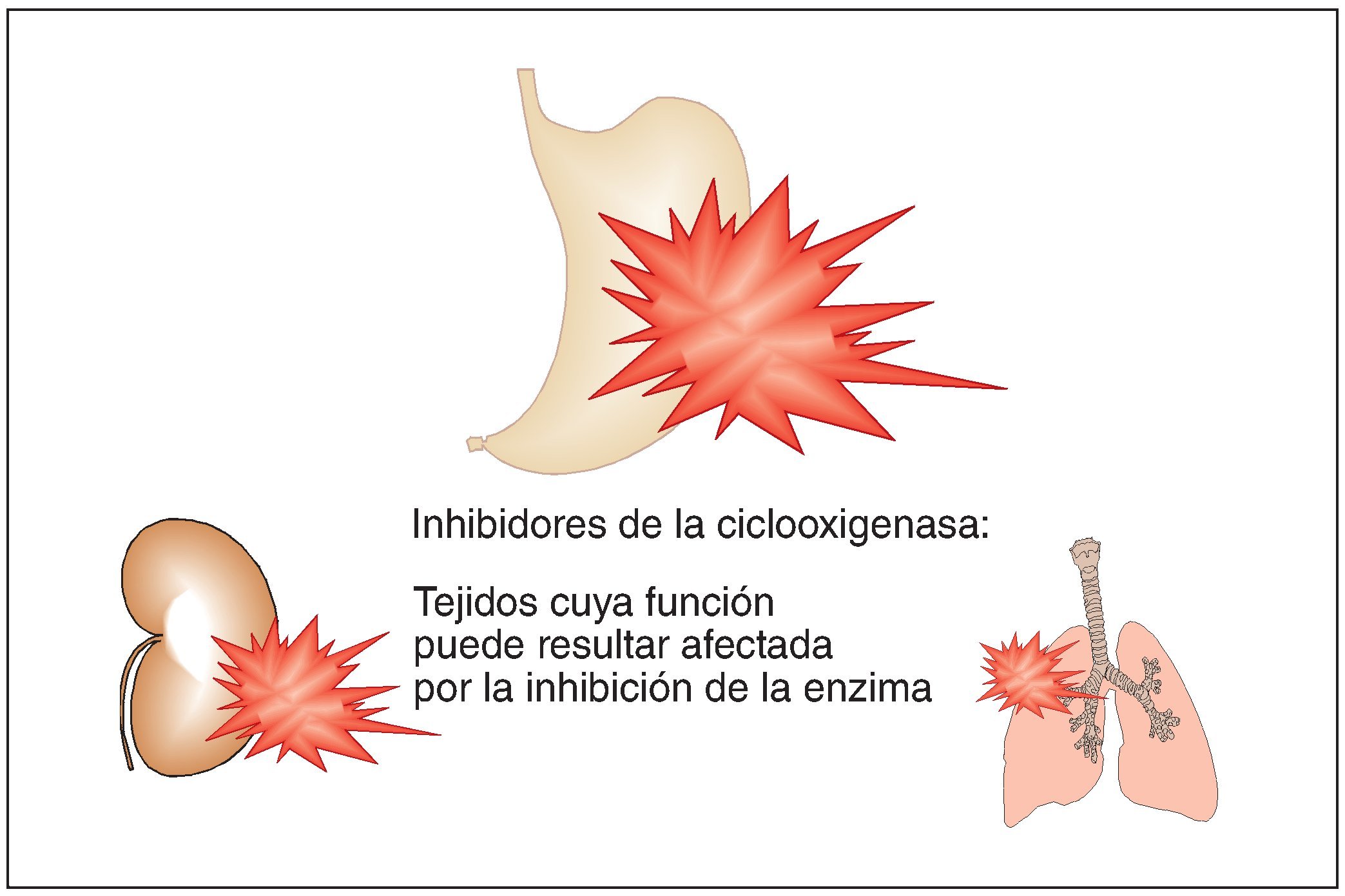
Figura 2. Estructuras orgánicas cuya función puede quedar afectada por los analgésicos del grupo de los inhibidores de la ciclooxigenasa (antiinflamatorios no esteroideos, AINE).
Irritación/úlceras de la mucosa gastrointestinal.
Alteraciones de la perfusión renal. En condiciones de hipovolemia, la perfusión renal depende de forma crítica de las prostaglandinas. El organismo reacciona a un estado de hipovolemia con una vasoconstricción que debe mantener la presión necesaria para la perfusión. Esta vasoconstricción se produce, entre otros mecanismos, a través de una activación del sistema reninaangiotensina. En el riñón, la angiotensina da lugar a una liberación de prostaglandinas que antagonizan funcionalmente los efectos de la angiotensina sobre los vasos renales. De este modo se asegura la perfusión esencial para el riñón aún en condiciones de hipovolemia.
Disnea. En caso de hipoventilación pulmonar, el organismo reacciona con una liberación de prostaglandinas que provocan una dilatación de las vías respiratorias. Los inhibidores de la ciclooxigenasa pueden desencadenar una crisis asmática en pacientes con predisposición a la broncoconstricción.
Embarazo. Las prostaglandinas favorecen el cierre del conducto arterioso hacia el final del embarazo y desempeñan un papel destacado en la inducción y el mantenimiento de las contracciones de parto. Su uso está contraindicado en la última fase del embarazo. Dado que no se puede descartar totalmente un efecto embriotóxico, debe evaluarse cuidadosamente la relación beneficioriesgo antes de cada nueva prescripción, independientemente de la fase del embarazo.
Los efectos adversos expuestos son comunes a todos los inhibidores de la cicolooxigenasa y explican las contraindicaciones siguientes:
úlcera gástrica, antecedentes de úlcera
hipovolemia (sistema renina-angiotensina-aldosterona activado)
tendencia al broncospasmo, asma
tratamiento con ácido acetilsalicílico a dosis bajas (profilaxis de la trombosis arterial, profilaxis del infarto y reinfarto)
En lo que se refiere a la última contraindicación hay que advertir que toda inhibición de las ciclooxigenasas, ya sea a través de inhibidores específicos o de inhibidores selectivos de la COX-2, neutraliza el efecto antiagregante del ácido acetilsalicílico. Dado que se utiliza el ácido salicílico a dosis bajas para la prevención del infarto de miocardio, es posible que la neutralización de su efecto provoque un aumento de acontecimientos adversos graves (véase el artículo «Ácido acetilsalicílico: inhibición de la agregación plaquetaria» en este mismo número).
Criterios de elección en la prescripción de inhibidores de la ciclooxigenasa
En el pasado han abundado los intentos de clasificar los distintos inhibidores de la ciclooxigenasa en función de la probabilidad de desencadenar este tipo de efectos adversos. Sin embargo, los resultados obtenidos no han permitido establecer un orden concreto. Existe la posibilidad de inhibir selectivamente la ciclooxigenasa inducible (COX-2) protegiendo al mismo tiempo la ciclooxigenasa constitucional (COX-1). Los inhibidores selectivos de la COX-2 (celecoxib, etoricoxib, parecoxib) tienen una menor capacidad ulcerogénica, si bien no están totalmente exentos de este riesgo.
Para algunas sustancias concretas se han descrito efectos adversos sobre la presión arterial, el hígado, la piel y el hemograma, observados sobre todo en los tratamientos sintomáticos a largo plazo con estos principios activos de la artritis reumatoide o las fases activas de la artrosis. Por lo tanto, no se pueden utilizar los efectos adversos para distinguir los principios activos si se administran en tratamientos analgésicos a corto plazo.
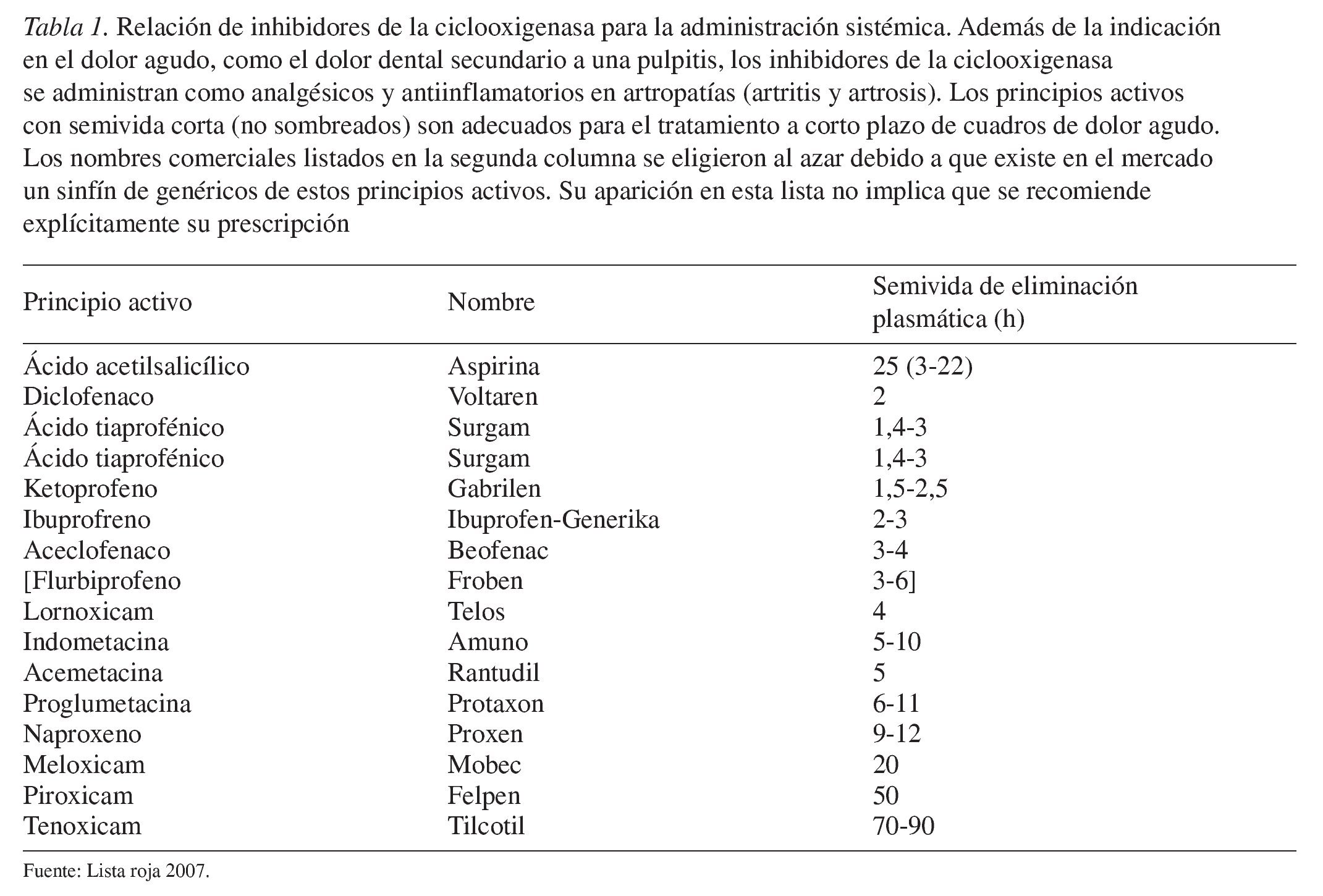
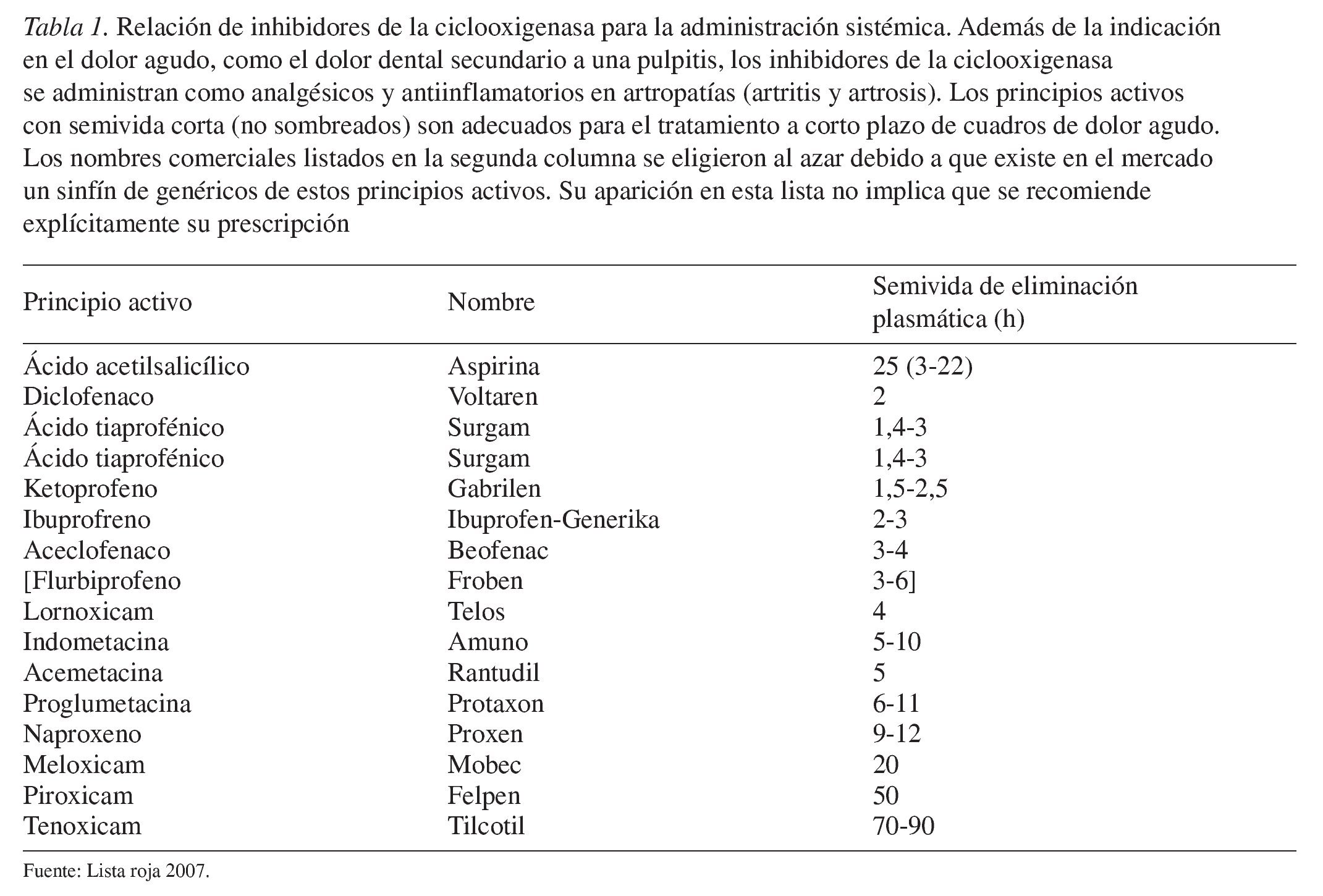
Los principios activos se pueden ordenar en función de su permanencia en el organismo (semivida de eliminación plasmática) (tabla 1). Los principios activos con una semivida de eliminación superior a 4 h no son adecuados para el tratamiento a corto plazo del dolor agudo.
Las características especiales del ácido acetilsalicílico
El ácido acetilsalicílico es un principio activo especial. Es el único principio activo que provoca una inhibición irreversible de la ciclooxigenasa. Esta peculiaridad es la que permite la inhibición selectiva de la ciclooxigenasa en las plaquetas en condiciones de administración regular a dosis bajas. Administrado regularmente a dosis bajas, el ácido acetilsalicílico es un inhibidor de la agregación plaquetaria. Este efecto se aprovecha frecuentemente en la profilaxis del infarto de miocardio y en menor medida (por su menor eficacia) en la prevención del ictus. A dosis bajas, el ácido acetilsalicílico inhibe la agregación y la adhesión de las plaquetas por lo que disminuye el riesgo de trombosis en las arterias coronarias (lesión del endotelio vascular, agregación plaquetaria, obstrucción coronaria). El principio activo no influye en la coagulación sanguínea ni a dosis antiagregantes (30-100 mg/día) ni a dosis analgésicas (500-1.000 mg 3x/día). Los niveles de protrombina no disminuyen hasta dosis muy altas.
La biodisponibilidad limitada confiere otra característica especial al ácido acetilsalicílico. El principio activo es un éster que, después de su administración oral, se descompone parcialmente ya en la luz intestinal, pero a más tardar en el momento de su paso a la sangre portal y a su paso por el hígado (fig. 3). Los productos de degradación, ácido acético y ácido salicílico, carecen de efecto a las concentraciones generadas incluso a dosis terapéuticas muy altas. En condiciones óptimas, el 25% de la dosis de ácido acetilsalicílico ingerida por vía oral llega a la circulación mayor y pasa a los tejidos para ejercer su efecto analgésico. Sin embargo, en situaciones de dolor las condiciones de absorción no son en absoluto las óptimas. Se ha demostrado que el dolor prolonga el vaciado gástrico y con ello ralentiza el tránsito del contenido gástrico hasta las superficies intestinales en las que tiene lugar la absorción.
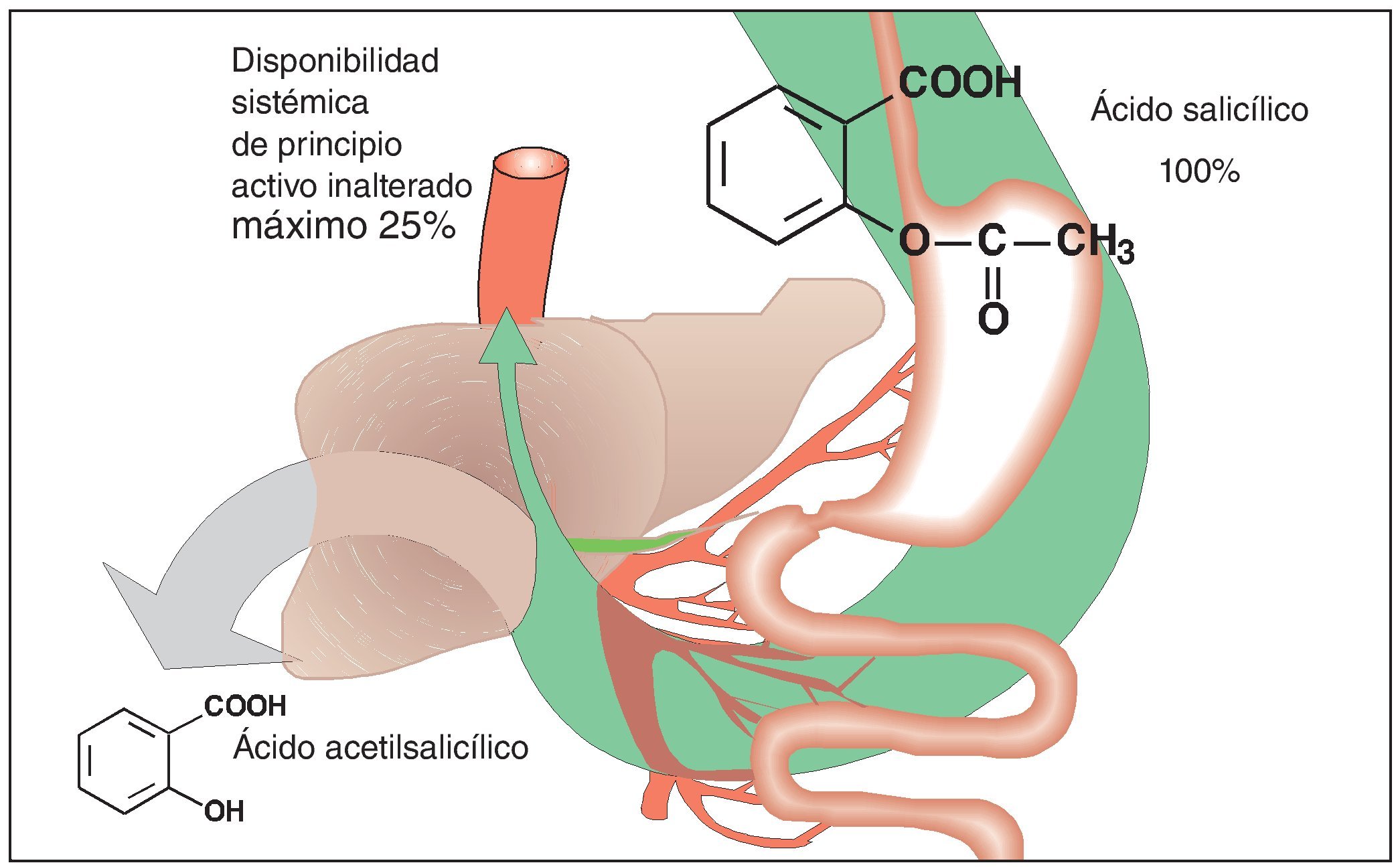
Figura 3. El ácido acetilsalicílico es absorbido en el intestino delgado después de su administración oral. Después de su paso a la circulación sistémica (vena porta) y a su paso por el hígado, el ácido acetilsalicílico se descompone en ácido salicílico y ácido acético (eliminación presistémica). El retraso en la disponibilidad del principio activo en forma disuelta a nivel del epitelio intestinal donde tiene lugar la absorción provoca forzosamente una disminución de la cantidad de principio activo inalterado disponible a nivel sistémico.
Todos los principios activos analgésicos sufren las consecuencias del vaciado gástrico lento en situaciones de dolor, pero especialmente el ácido acetilsalicílico debido a su inestabilidad química. La prescripción de un comprimido efervescente es una buena solución a este fenómeno. De este modo el paciente ingiere el principio activo en una solución de bicarbonato sódico, lo que favorece su disponibilidad más rápida en las superficies intestinales. La mayoría de los principios activos están disponibles en forma de comprimidos efervescentes (tabla 2).

Paracetamol
Se desconoce el mecanismo de acción analgésico de paracetamol. Existen indicios de que el paracetamol reacciona con ácido araquidónico después de la desacetilación a 4-aminofenol y que el producto obtenido inhibe la síntesis de prostaglandinas en el sistema nervioso central. La ausencia de esta descomposición a 4-aminofenol a nivel periférico podría explicar el motivo por el que el paracetamol carece de los efectos adversos característicos de los inhibidores de la ciclooxigenasa. La mención del posible mecanismo de acción parece importante, ya que puede ser útil para decidir posibles alternativas terapéuticas en los casos en que los inhibidores de la ciclooxigenasa no proporcionan una analgesia suficiente. Por lo tanto, en base al supuesto mecanismo de acción tendría poco sentido sustituir un ibuprofeno por paracetamol.
Hasta ahora se ha considerado que el paracetamol tiene una muy buena tolerabilidad. No se ha conseguido documentar para paracetamol un efecto nefrotóxico como el observado después de la ingesta excesiva a largo plazo de fenacetina, un compuesto químicamente muy parecido a paracetamol (lo que posiblemente también se debe al hecho de que el número de personas que abusan del consumo de paracetamol fue notablemente inferior al observado para fenacetina). Sin embargo, está fuera de toda duda el efecto hepatotóxico de la ingesta de dosis únicas superiores a 10 g. El mecanismo del efecto hepatotóxico está perfectamente establecido (fig. 4). Después de la administración de dosis terapéuticas, el paracetamol se une a ácido glucurónico o a ácido sulfúrico en el hígado. Los productos de unión se eliminan rápidamente por vía renal. Una fracción mínima de paracetamol es oxidada en el hígado para formar quinonimina. Se trata de una molécula altamente reactiva que es inactivada a través de su unión a glutatión hepático. En caso de dosis de paracetamol superiores a 10 g, los mecanismos de unión quedan saturados, y los excedentes de paracetamol pasan a la vía oxidativa para formar quinonimina. Las reservas de glutatión son insuficientes para neutralizar el agente reactivo. Éste reacciona con las proteínas del hepatocito y provoca su destrucción. Por lo tanto, la administración de dosis altas de paracetamol provoca previsiblemente una insuficiencia hepática. La detección precoz permite prevenir la insuficiencia hepática inexorable mediante la administración de aminoácidos azufrados (con grupos SH, como n-acetilcisteína), es decir, de análogos del glutatión.
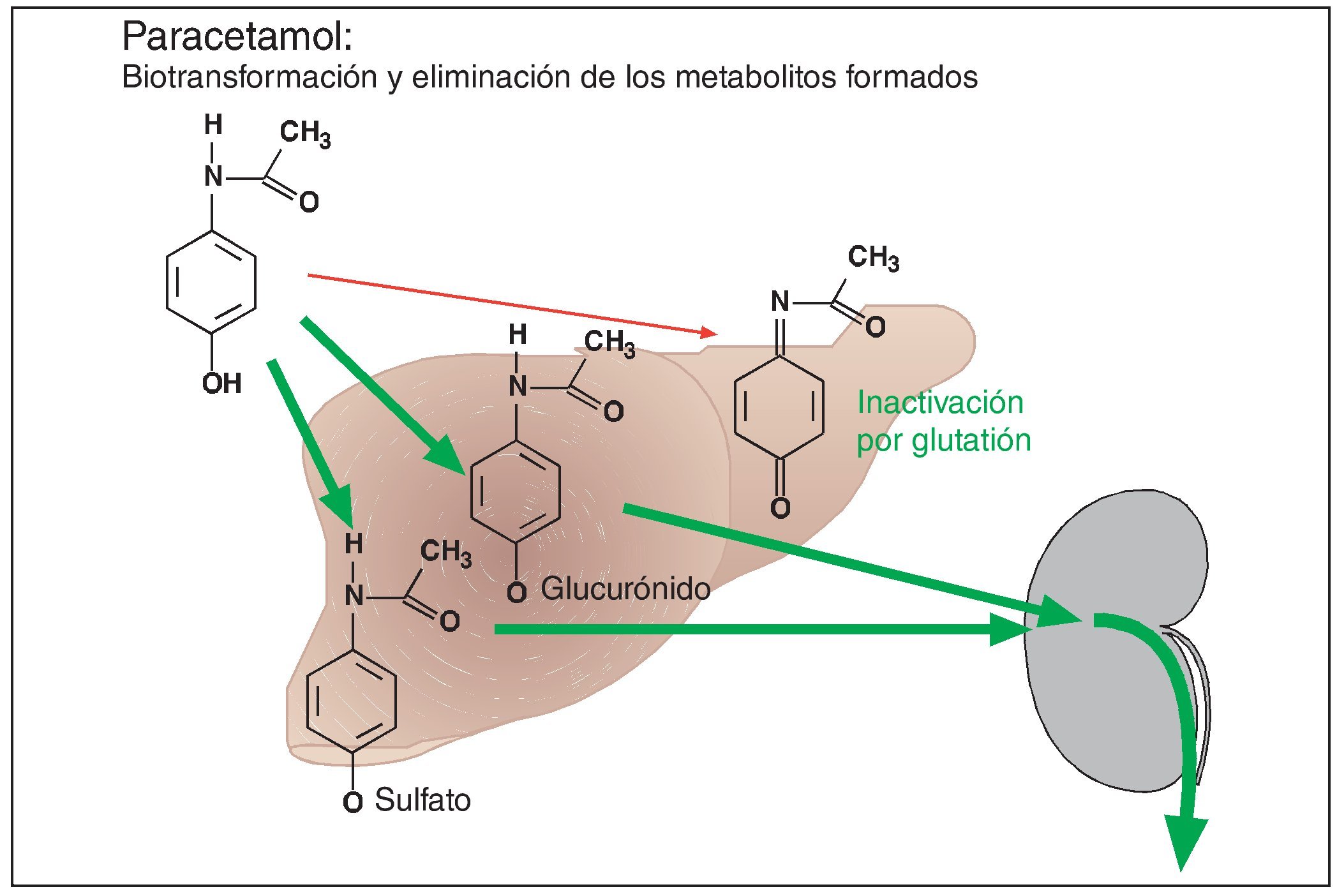
Figura 4. Después de su absorción, el paracetamol se une en el hígado a ácido glucurónico o a ácido sulfúrico mediante un proceso catalizado por enzimas. Los productos de reacción ácidos se eliminan por vía renal mediante un proceso de transporte activo. Una pequeña parte de este principio activo es oxidada en el hígado a quinonimina. Este metabolito reactivo es inactivado por glutatión. El efecto hepatotóxico de la quinonimina sólo se puede manifestar en caso de agotamiento de las reservas hepáticas de glutatión. Esta situación se da después de la administración de dosis individuales altas de paracetamol (> 10 g), ya que las enzimas de unión quedan desbordadas, desviándose concentraciones elevadas de paracetamol a la vía oxidativa para la formación de quinonimina hepatotóxica.
En 2006 se publicaron los resultados de un estudio en el que personas sanas ingirieron 1.000 mg de paracetamol cada 6 horas durante 14 días. En estas condiciones, se observó un aumento transitorio de la concentración plasmática de la enzima alanina aminotransferasa (ALT ~ ALAT ~ GPT) hasta valores medios superiores al doble (aproximadamente 100 U/l) del valor de referencia (50 U/l). El aumento de su concentración se considera como un indicador de daño celular hepático. Algunos investigadores opinan que estos resultados confirman la hepatotoxicidad de paracetamol a dosis terapéuticas y desplazan la relación beneficio-riesgo hasta el punto de que su prescripción ya sólo está justificada en casos excepcionales. Sin embargo, los autores del estudio sostienen que la existencia de una relación entre el aumento de la ALT y la ingesta de paracetamol es importante, ya que en el marco del estudio diagnóstico de un aumento de la ALT puede evitarse la búsqueda laboriosa de otras causas de hipertransaminasemia de obtener en la anamnesis una respuesta afirmativa a la pregunta acerca de un posible tratamiento con paracetamol. Se deberían esperar los resultados de otros estudios antes de eliminar el paracetamol del arsenal de analgésicos considerados seguros.
Metamizol
El metamizol posee propiedades analgésicas y antipiréticas. No se conoce el mecanismo de acción de metamizol. No provoca efectos adversos característicos que pudieran proporcionar indicios acerca de posibles lugares de acción. El metamizol, introducido en el mercado hace 90 años, gozaba hasta hace 35 años de gran popularidad en Alemania y también en otros países, dado que alivia eficazmente el dolor visceral y el dolor de tipo cólico. Se sabe desde hace tiempo que la administración intravenosa de metamizol puede desencadenar un estado de shock o reacciones alérgicas localizadas, aunque es probable que el riesgo de provocar un colapso cardiocirculatorio dependa de la velocidad de inyección. Este tipo de reacciones no se observa o sólo se observa en casos aislados después de la administración oral. Las alertas en relación con un posible efecto adverso en forma de agranulocitosis provocaron reacciones de preocupación entre la clase médica (disminución de las prescripciones) y también en las autoridades reguladoras (paso al régimen de dispensación con receta médica). Los estudios epidemiológicos iniciados a raíz de estas alertas no pudieron descartar totalmente una relación entre la ingesta de metamizol y una afectación grave de los leucocitos. Sin embargo, dada la excepcionalidad de este efecto adverso no se logró cuantificar el riesgo.
Metamizol es un analgésico eficaz y de perfil único, por lo que debe quedar a disposición de los pacientes como analgésico de reserva. De cara a la administración se debe tener en cuenta la posible afectación del hemograma, lo que permitirá reaccionar con rapidez ante signos incipientes de alteración del recuento de granulocitos. Este procedimiento proporciona buenos resultados como se ha podido comprobar con otros medicamentos que presentan el mismo riesgo como, por ejemplo, el antipsicótico clozapina.
Los datos presentados muestran, ya sólo por sus efectos analgésicos específicos, que metamizol es un analgésico poco adecuado para cuadros de dolor dental.
Opioides
Bajo el término opioides se agrupan aquellos principios activos que, igual que la morfina, interaccionan con un lugar de acción determinado en el organismo, es decir, con un receptor (farmacológico). En este caso, el receptor es un receptor opioide. Los intentos de clasificar el grupo de los opioides en función de su afinidad por los subtipos de receptores opioides no han proporcionado una clasificación útil a efectos prácticos.
Los receptores opioides están distribuidos tanto en el sistema nervioso central como en el sistema nervioso periférico. Su activación por acción de un opioide disminuye la excitabilidad neuronal. Se cree que el efecto analgésico es el resultado de efectos espinales y también de efectos supraespinales. Si se explotan todas las posibilidades, los opioides pueden llegar a proporcionar una analgesia que permite llevar a cabo intervenciones quirúrgicas sin anestesia total. Sin embargo, en estas condiciones se produce una disminución considerable del estado de alerta y una depresión del centro respiratorio. Para el uso en la anestesia, se dispone de opioides especiales, de acción corta y de fácil manejo.
En el tratamiento a largo plazo de cuadros de dolor o para el tratamiento sintomático del dolor agudo en el paciente ambulatorio son útiles los opioides de liberación retardada y cuya eliminación permite alcanzar concentraciones plasmáticas suficientes durante varias horas. Para este tipo de tratamiento se dispone, además de la morfina, de los principios activos indicados en la tabla 3. Los opioides se han utilizado y se siguen utilizando a menudo en el tratamiento del dolor oncológico. Para los no entendidos estos principios activos llevan asociado a menudo el estigma de ser un analgésico reservado para los estadios terminales de un cáncer.

Todas las sustancias de este grupo tienen el inconveniente de ser adictivas y de entrañar un riesgo importante de consumo abusivo con consecuencias graves. Esta problemática fue uno de los motivos por los que se impuso una prescripción altamente restrictiva en el pasado. Sin embargo, la situación cambió sustancialmente con la introducción de preparados de liberación retardada, dado que esta forma farmacéutica presenta un riesgo nulo o como mínimo mucho menor de provocar adicción a largo plazo. Hoy en día impera la teoría de que la administración de dosis analgésicas de opioides en las indicaciones autorizadas no provoca dependencia.
Otros efectos adversos de los opioides incluyen:
Disminución de la capacidad de respuesta del centro respiratorio
Estreñimiento
Náuseas y vómitos
La depresión del centro respiratorio es un efecto adverso frecuente asociado al uso de los opioides en la anestesia, pero no se observa en tratamientos con preparados de liberación retardada en pacientes ambulatorios. Es posible que el aumento progresivo de las concentraciones plasmáticas del principio activo permita una adaptación del centro respiratorio. Sin embargo, no se debería olvidar la posibilidad de la depresión respiratoria, dado que los opioides están contraindicados en pacientes con enfisema pulmonar.
Los opioides provocan estreñimiento siempre, con una asiduidad que obliga al uso preventivo de laxantes en los tratamientos analgésicos a largo plazo.
Los vómitos son un efecto adverso temido de los opioides administrados en la anestesia, pero son menos frecuentes después de la administración de preparados de liberación retardada. Sin embargo, éstos se asocian frecuentemente a náuseas. El paciente considera las náuseas como un efecto adverso desagradable, pero las tolera porque suelen coincidir con el inicio del efecto analgésico.
La codeína y el tramadol no llevan asociado el estigma del analgésico potente para los dolores del cáncer en fase terminal y se pueden utilizar en el paciente reticente por eventuales trabas psicológicas para el tratamiento del dolor no canceroso. La codeína ocupa una posición particular entre los opioides que no puede explicarse con argumentos farmacológicos, pero que se ve reforzada por el hecho de que, en las presentaciones habituales, no está sujeta a la prescripción y dispensación con receta de estupefacientes.
Tramadol tampoco está sujeto a la prescripción y dispensación con receta de estupefacientes. Tramadol es un opioide menos eficaz. Se sospecha que una segunda propiedad farmacológica, la inhibición de la recaptación de noradrenalina, también contribuye a su efecto analgésico. Esto podría potenciar la inhibición de la unión sináptica a nivel medular descrita para los opioides. Se ha demostrado la presencia de receptores α2-adrenérgicos en las sinapsis del asta posterior, cuya mayor activación podría desencadenar el supuesto efecto adicional de tramadol. No se dispone de evidencias contrarias a la prescripción de codeína o tramadol para el tratamiento del dolor dental en casos en que los inhibidores de la ciclooxigenasa fracasan o proporcionan una analgesia insuficiente.
Flupirtina y ziconotida
La flupirtina no es un opioide ni un inhibidor de la ciclooxigenasa. Carece de propiedades antiinflamatorias y antipiréticas, pero es un relajante muscular. Los efectos adversos incluyen cansancio, mareo y trastornos gastrointestinales. No se dispone de datos que apoyen el uso de flupirtina en el tratamiento del dolor dental.
La ziconotida es un análogo de un veneno (ω-conotoxina) del caracol de mar Conus magus. Este principio activo inhibe de forma altamente específica un canal de calcio neuronal y debe ser administrado por vía intradural. Es un analgésico no apto para el paciente ambulatorio ni para su uso en odontología debido a su vía de administración, pero sobre todo por sus efectos adversos centrales graves.
Estos dos principios activos se han mencionado aquí únicamente para completar la gama de analgésicos.
Preparados combinados analgésicos
En el pasado se produjeron debates, en ocasiones encendidos, en torno a unos analgésicos de uso frecuente que contenían más de un principio activo analgésico, en muchos casos también en combinación con un sedante/hipnótico y/o cafeína. El rechazo contundente de preparados combinados que contenían un sedante/hipnótico estaba y sigue estando justificado desde el punto de vista farmacológico y, afortunadamente, han sido retirados del mercado. Sin embargo, en este momento es más difícil encontrar argumentos concluyentes en contra de los preparados combinados todavía disponibles en el mercado. Parece resultar difícil documentar e incluso cuantificar el beneficio de la adición de cafeína. Algunos estudios muestran la posibilidad de disminuir la dosis individual eficaz mediante la adición de cafeína. Sin embargo, no se sabe en qué beneficia al paciente la disminución de esta dosis.
A los partidarios de los monopreparados, es decir, a aquellos que rechazan la combinación de principios activos analgésicos, como el ácido acetilsalicílico y el paracetamol, se les podría responder que la mayoría de los analgésicos poseen probablemente más de un mecanismo de acción y que, por lo tanto, los monopreparados también generan una mezcla de efectos.
Dolor neuropático
El dolor neuropático tiene su origen en una alteración del sistema nociceptivo. En el ámbito de la odontología pueden producirse lesiones nerviosas a consecuencia de fracturas, de una intervención quirúrgica, una inyección o de una infección. Las compresiones nerviosas pueden ser secundarias a un implante, al crecimiento óseo o a una masa tumoral.
El dolor neuropático puede provocar los síntomas siguientes:
Odontalgia atípica
Dolor fantasma
Dolor dental u oral persistente
Ardor bucal y lingual idiopático
El dolor neuropático forma parte de los cuadros de dolor crónico. A diferencia de lo que ocurre en el dolor nociceptivo patológico, los tratamientos farmacológicos del dolor neuropático no proporcionan resultados inmediatos. El paciente y el médico deben tener claro desde el principio que el tratamiento no será fácil ni rápido. Los principios activos del grupo de los anticonvulsionantes/ antiepilépticos y antidepresivos han mostrado ser eficaces en el tratamiento del dolor neuropático.
Como se ha mencionado antes, el patrón de las proteínas funcionales de los nervios sensitivos se modifica como consecuencia del daño nervioso. El exceso de excitabilidad resultante puede tratarse eficazmente con principios activos que inhiben sobre todo descargas eléctricas de alta frecuencia. Esto explica en parte los buenos resultados conseguidos con pruebas terapéuticas en las que se han utilizado anticonvulsionantes como carbamacepina (Tegretal, Timonil, entre otros) o con lamotrigina (Lamictal). Con la administración de gabapentina (Neurontin), pregabalina (Lyrica) y amitriptilina (Saroten, entre otros) se pretende disminuir la excitabilidad sináptica y la eficiencia con la que se propaga la excitación eléctrica, por ejemplo en el asta posterior, entre la primera y la segunda neurona aferente.
El neurólogo es el médico especializado en el tratamiento de dolores neuropáticos, como los observados fuera del ámbito de la odontología. Ante un paciente odontológico con dolor neuropático se debería considerar la posibilidad de su derivación a un centro especializado en el tratamiento de dolores neuropáticos, debido a la multiplicidad de medidas farmacológicas disponibles y a que al menos algunas de ellas se pueden acompañar de efectos adversos importantes.
Nos parece importante insistir en el hecho de que el tratamiento del dolor neuropático es un proceso largo y que exige paciencia y perseverancia tanto por parte del paciente como del médico. Sería frívolo dejar entrever al paciente que se obtendrán resultados terapéuticos a corto plazo.
Correspondencia: A. Ziegler.
Richterstrabe 18a, 24159 Kiel, Alemania.
Correo electrónico: aziegler@pharmakologie.uni-kiel.de