





INTRODUCCION
El síndrome de Swyer es una disgenesia gonadal pura, con cariotipo 46XY, fenotipo femenino normal y ausencia completa de tejido gonadal funcionante. Los genitales internos son normales, salvo la presencia de cintillas gonadales bilaterales. La forma de presentación clínica más frecuente es amenorrea primaria, aunque también puede debutar con dolor y distensión abdominal, con o sin hemoperitoneo, cuando existen tumores germinales asociados, como es el caso que presentamos. Pueden existir alteraciones genéticas, en el gen SRY u otros, pero sólo se han descrito en un 20% de casos. El riesgo de neoplasia gonadal es alto, entre un 25-30%; el disgerminoma y el gonadoblastoma son las más comúnmente asociadas.
CASO CLÍNICO
Paciente de 13 años de edad, sin antecedentes personales o familiares de interés. Estadio de Tunner P2-3 y T4, sin menarquia, talla de 1,77 cm y peso 63 kg, que consulta por aumento del perímetro abdominal con dolor. Se describe, tras el estudio de imagen, una tumoración sólida de 13 cm, con evidentes calcificaciones, dependiente de anejo derecho; ascitis a tensión. No presenta adenopatías (fig. 1). En estudios analíticos presenta BHCG de 94,9 mU/ml y perfil hormonal prepúber. Se practicó una laparotomía urgente y se halló una tumoración ovárica derecha de 10 cm, con una cápsula íntegra. El útero y el anejo izquierdos eran normales para edad de la paciente. El diagnóstico intraoperatorio fue tumor de los cordones sexuales-estroma sospechoso de células Sertoli-Leyding. Se le practicó una anexectomía derecha, una omentectomía, una apendicectomía y una biopsia de ovario contralateral más lavados peritoneales. El resultado definitivo de anatomía patológica fue de disgerminoma (fig. 2) para el ovario derecho, gonadoblastoma (fig. 3) para la biopsia del izquierdo, y el líquido peritoneal fue negativo a células malignas. La paciente recibió tratamiento adyuvante con 4 ciclos de bleomicina, etopósido y cisplatino. En la revisión posquimioterapia en nuestra consulta la paciente presentaba: fenotipo femenino normal, genitales externos normales, himen íntegro, vagina de 9 cm de longitud. Los estudios de imagen y los marcadores tumorales analíticos fueron normales, incluido el BHCG. El perfil gonadal prepúber. El estudio del cariotipo desveló un 46XY con gen SRY sin alteraciones, lo cual motivó nuestra propuesta para la gonadectomía izquierda, que se realizó por laparoscopia, y se encontró un resultado anatomopatológico de cintilla gonadal normal. La paciente comenzó un tratamiento con estroprogestágenos cíclicos. Actualmente, se encuentra eumenorreica, libre de enfermedad y con fenotipo femenino P4-T4.
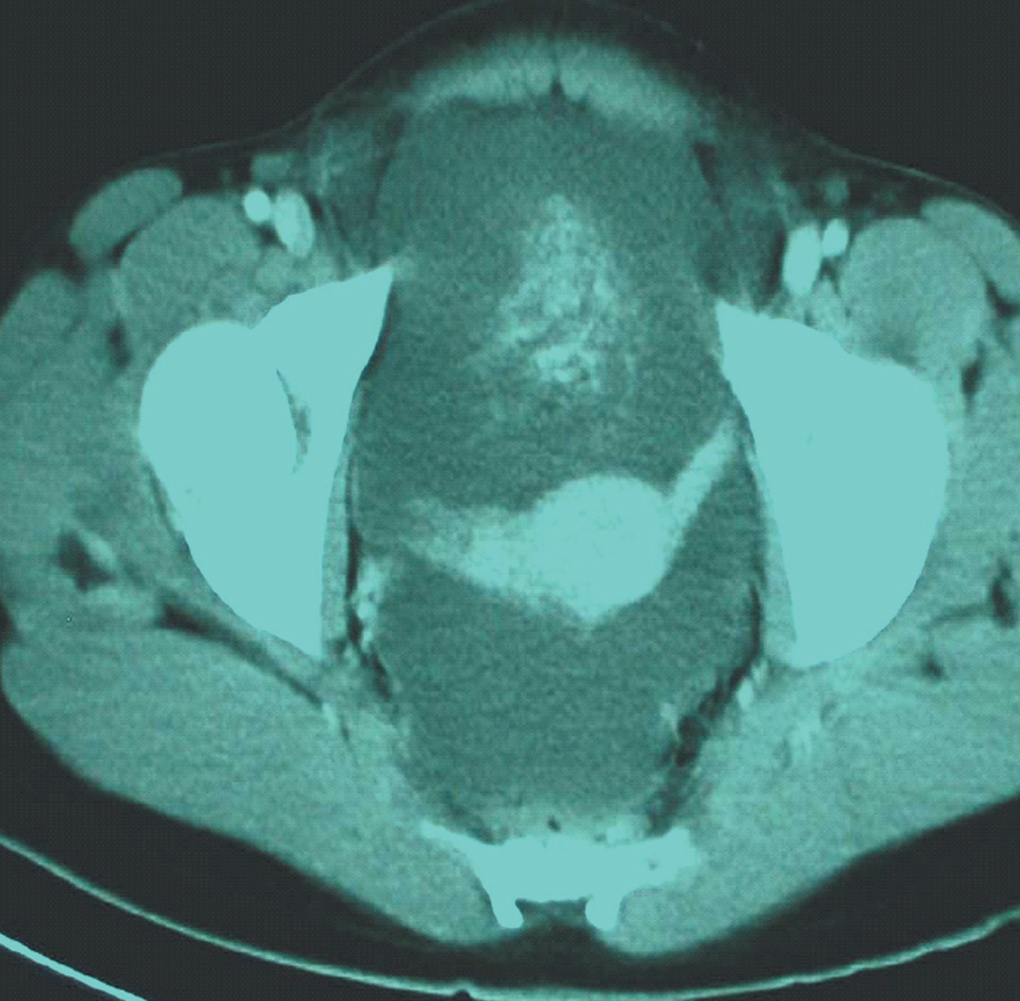
Figura 1. Tumoración pélvica.
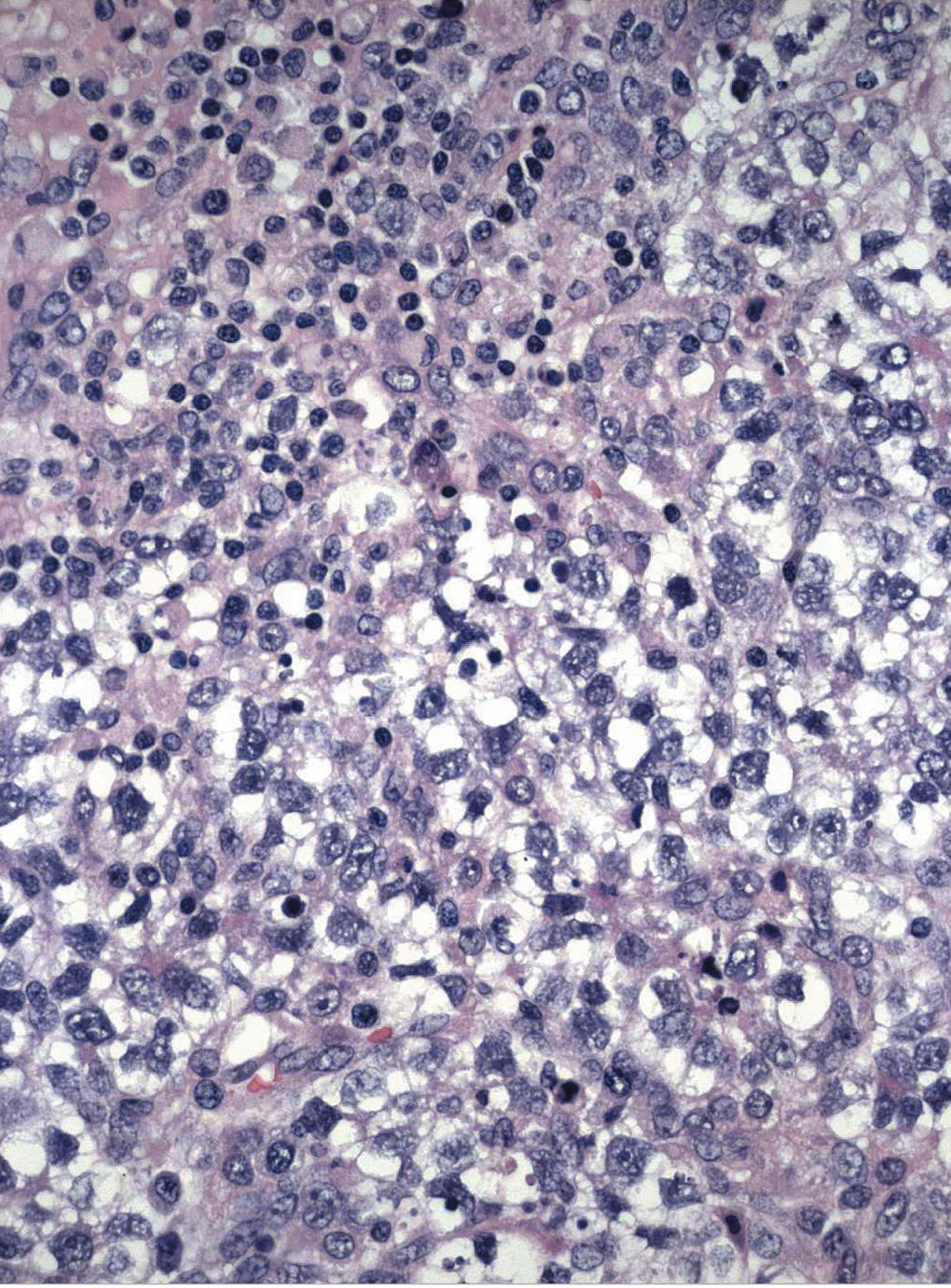
Figura 2. Disgerminoma.
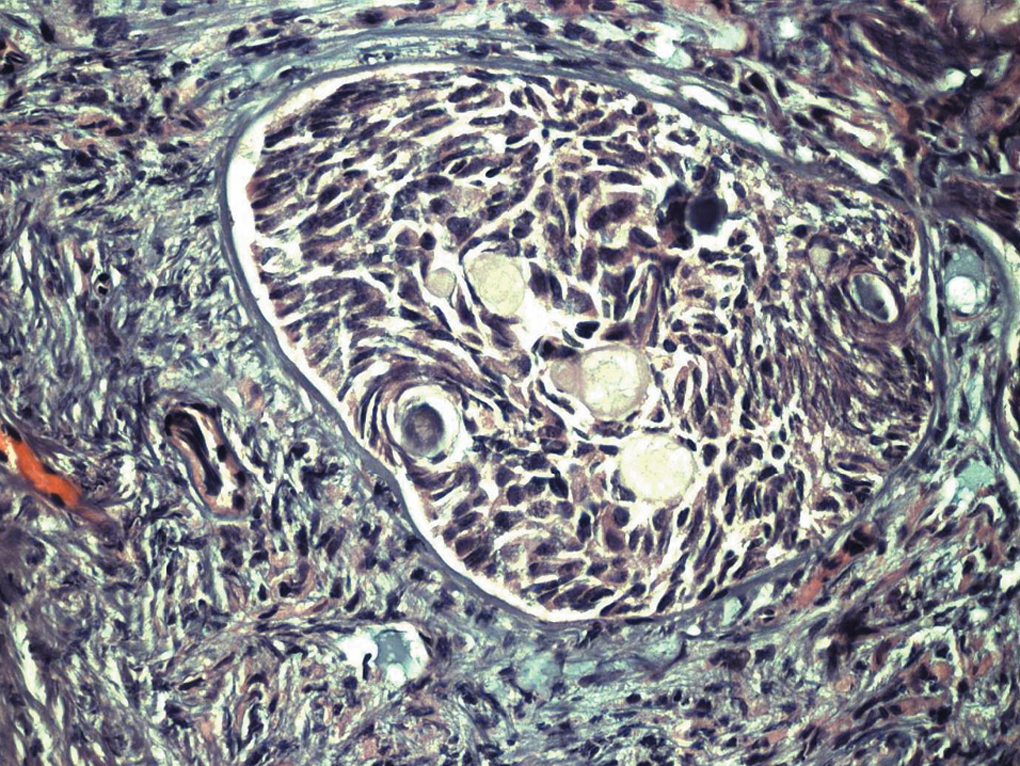
Figura 3. Gonadoblastoma.
DISCUSION
En 1955, Swyer documentó los 2 primeros casos de disgenesia gonadal pura en su artículo titulado: "Seudohermafroditismo masculino: una forma no descrita hasta ahora". Sus pacientes presentaban fenotipo femenino, y el motivo de consulta era amenorrea primaria. El fenotipo se caracterizaba por: estatura normal, hábito eunucoide, escaso o nulo desarrollo mamario, vello púbico y axilar, vagina y cérvix de características normales, útero pequeño e incapacidad para identificar anejos. El estudio del cariotipo de ambas pacientes reveló un 46XY. Ninguna de ellas presentaba estigmas de síndrome de Turner. A pesar del enorme interés científico que suponía el conocimiento del estado histológico de las gónadas, Swyer1 estableció que la laparotomía no estaba justificada. En nuestro caso, la paciente presentaba un desarrollo mamario normal, ausencia de hábito eunucoide y el motivo de consulta fue dolor abdominal.
El concepto de disgenesia gonadal pura hace referencia a una variante específica con un defecto en la organogénesis en la cual las pacientes fenotípicamente femeninas, con cariotipo 46XY, presentan ausencia completa de tejido gonadal funcionante. Este tejido está anatómicamente compuesto por unas cintillas gonadales rudimentarias, alargadas, bilaterales y no funcionantes, compuestas por tejido fibroso. El desarrollo de los genitales internos femenino es normal2.
Las cintillas gonadales bilaterales están siempre presentes en las formas completas, mientras que en las formas parciales pueden presentar distintos grados de virilización, dependiendo de la gravedad en la afectación testicular. Una posible explicación para esta afección son las mutaciones en el gen SRY, que producirían alteraciones en la diferenciación testicular. El gen SRY (localizado en la proximidad de la región seudoautosómica del brazo corto del cromosoma Y) se considera como el primer eslabón de una cascada de fenómenos que conducen a la diferenciación testicular. El gen SRY tan sólo se expresa en el testículo durante el período de diferenciación gonadal. Ratones transgénicos a los que se ha manipulado el gen SRY han presentado desarrollo gonadal y fenotipo masculino, y algunas mujeres con cariotipo XY (síndrome de Swyer) presentan mutaciones en este gen que provocan su inactivación3. Sin embargo, sólo el 10-15% de las pacientes con la forma completa del síndrome presentan mutaciones en el gen, localizadas fundamentalmente en la región HMG. El resto de los casos probablemente se deban a: mutaciones de tipo autosómico, mutaciones ligadas al X (que estarían también involucrados en la cascada de diferenciación sexual), mutaciones en regiones del SRY aún sin identificar, u otras mutaciones que den lugar al mismo fenotipo4. Aunque el SRY fue identificado por primera vez hace 10 años, todavía sabemos increíblemente poco sobre su mecanismo de acción. Parece ser que modula una serie de complejas interacciones, que incluyen factores reguladores tanto positivos como negativos, los cuales estarían influidos por el ambiente celular, y esto conduciría a la expresión de genes masculinos específicos. A pesar de los avances más recientes, somos incapaces de explicar la causa genética de la mayoría de casos de disgenesia gonadal 46XY5.
Sin embargo, existen descritas varias alteraciones genéticas en la bibliografía médica que se han relacionado con el Síndrome de Swyer. En 1997, MacDonald et al6 describen un caso de disgenesia gonadal y gonadoblastoma en una paciente 46XY con una deleción del 9p24, relacionada con una traslocación familiar. El cromosoma 9 alterado fue heredado del padre, el cariotipo de la paciente era 46XY, der(9)t(8;9)(p 21;p24)pat. Tras una revisión sobre el caso, observaron la asociación de la monosomía del brazo corto distal del cromosoma 9 con 46XY en 6 casos más. El hecho de que las 7 pacientes compartieran una deleción del brazo corto distal del cromosoma 9 coincide con la hipótesis de que la región 9p24 contiene uno o varios genes implicados en la determinación del sexo masculino.
Por otro lado, sabemos que entre el 10-30% de las pacientes con gónadas disgenéticas y cromosoma Y desarrollarán un tumor gonadal. Entre ellos, el gonadoblastoma y el disgerminoma son los más frecuentes.
En 1999, Uchara et al7 investigaron la relación entre la formación de estos tumores (gonadoblastoma y disgerminoma) en 3 pacientes con disgenesia gonadal pura XY, y su relación con aberraciones en el gen SRY. La paciente de 22 años, que no asociaba tumor, carecía de mutación o deleción en el SRY. En el tejido tumoral de las otras 2 pacientes se demostró por amplificación PCR (polimerase chain reaction), una secuencia específica en el cromosoma Y (DYZ1). Estos resultados sugieren que el SRY puede desempeñar un papel en la formación de tumores gonadales, especialmente disgerminoma.
La gonadectomía profiláctica, por tanto, está recomendada en estas pacientes, tan pronto el diagnóstico se confirme, a pesar de que estos tumores suelen desarrollarse en la infancia y adolescencia2,8,9. La vía de abordaje de elección es la laparoscópica, debido a su seguridad, buena relación coste-efectividad y a la recuperación postoperatoria más precoz10-12.
Debido al posible carácter hereditario de algunas formas de disgenesia gonadal XY, los miembros de la familia deben ser estudiados13.
Los valores hormonales no suelen verse alterados, con estrógenos normales o disminuidos, hormona folicoestimulante o folitropina (FSH) elevada o normal, y testosterona normal o discretamente elevada si existieran tumores secretores de andrógenos o estrógenos. Tras la gonadectomía o la extirpación de tumores, si los hubiera, el tratamiento consistirá en administrar estrógenos y progestágenos cíclicamente, para restaurar la menstruación y conseguir un desarrollo de caracteres sexuales secundarios adecuado14.
En cuanto al disgerminoma, el tratamiento más aceptado para estas pacientes es la estadificación adecuada con gonadectomía bilateral profiláctica, respetando el útero, para satisfacer el deseo genésico, ya que suelen ser pacientes jóvenes que podrían beneficiarse en un futuro de una fecundación in vitro (FIV) con donación de ovocitos. En los estadios Ib-IV debe instaurarse un tratamiento adyuvante habitualmente con quimioterapia combinada en regímenes como VBP (vinblastina, bleomicina y cisplatino) o BEP (bleomicina, etopósido y cisplatino)15.
Correspondencia:
Dra. A.I. Salazar Vera.
Servicio de Obstetricia y Ginecología.
Hospital Juan Ramón Jiménez.
Ronda Norte, s/n. 21005. Huelva. España.
Correo electrónico: f.palomeque@arrakis.es
Fecha de recepción: 4/02/04.
Aceptado para su publicación: 5/01/06.