




INTRODUCCION
Las trisomías parciales del brazo largo del cromosoma 1 son poco frecuentes, y en la mayoría de los casos la duplicación es el resultado de la segregación anormal en meiosis de una translocación equilibrada en uno de los progenitores. Habitualmente, se acompañan de malformaciones múltiples y se suelen establecer 2 grupos diferenciados de pacientes. Uno con graves malformaciones congénitas y corta vida media1, y otros con malformaciones más leves y mayor supervivencia2. Diferentes autores han intentado definir un síndrome asociado a esta cromosomopatía3.
La mayor parte de los casos descritos se refieren a trisomías parciales 1q32-qter4, que coinciden con el grupo de pacientes de malformaciones más leves, y su mayor incidencia puede indicar la existencia de un «sitio frágil» a ese nivel del cromosoma o bien que las roturas más proximales, al incluir un fragmento cromosómico más grande, sean incompatibles con la vida5. En este sentido, la duplicación 1q25-qter se considera como una condición letal a no ser que el segmento adicional esté translocado en un cromosoma X parcialmente inactivado6, en cuyo caso pueden presentar supervivencias de más de 1 año.
Nosotros exponemos un caso de trisomía parcial 1q por duplicación del segmento cromosómico 1q25-qter, diagnosticado prenatalmente, analizando las alteraciones ecográficas en la 16 semana de la gestación, así como los hallazgos del estudio necrópsico, y discutimos las similitudes y diferencias respecto a otros casos similares descritos en la bibliografía.
OBSERVACION CLINICA
Paciente de 31 años, que acude a la Unidad de Ecografía y Medicina Fetal del Hospital Universitario de Canarias para una evaluación gestacional rutinaria en la semana 16 cronológica gestacional. En la ecografía se detecta una gestación que biométricamente corresponde a 15 semanas, con un feto que muestra datos dismorfológicos consistentes en megavejiga urinaria con hipertrofia de paredes vesicales e hidronefrosis más visible en el riñón derecho, así como higroma quístico del cuello de 46 mm de sección (fig. 1). Dada la evidente relación del cuadro con una cromosomopatía, se practica amniocentesis.
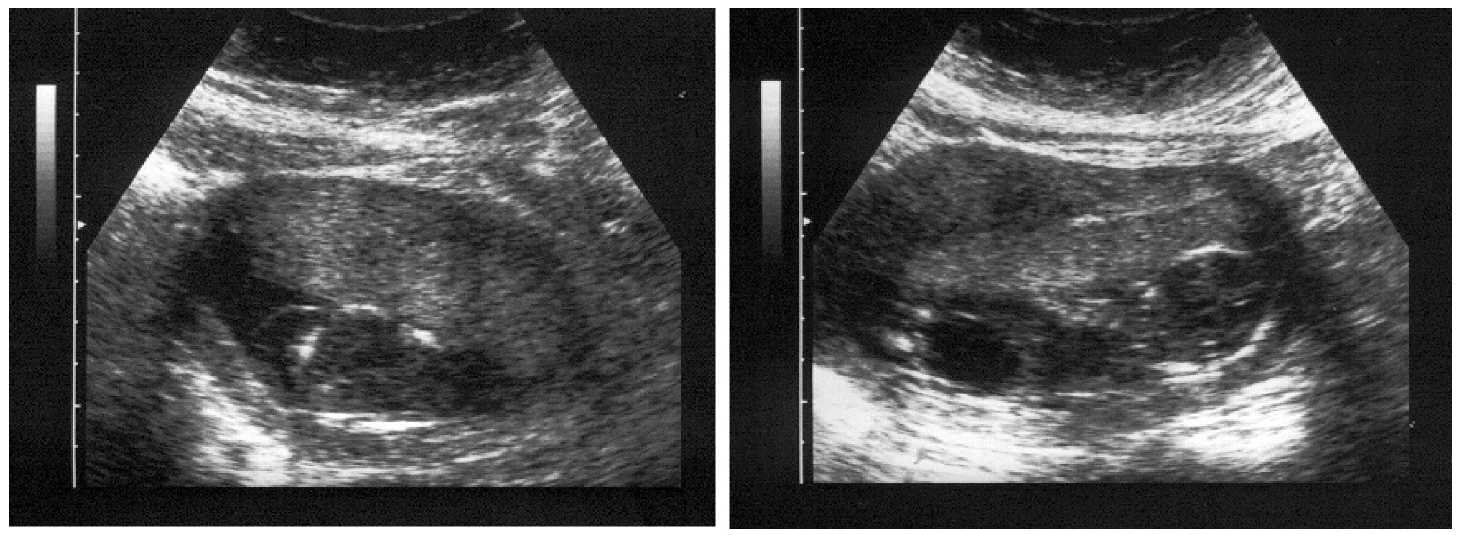
Figura 1.Hallazgos ecográficos destacados (16 semanas). A) Higroma quístico. B) Megavejiga.
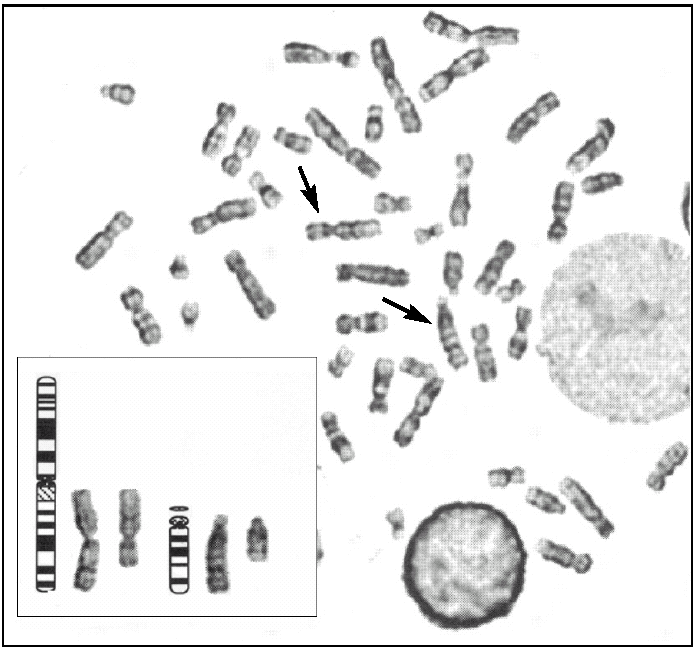
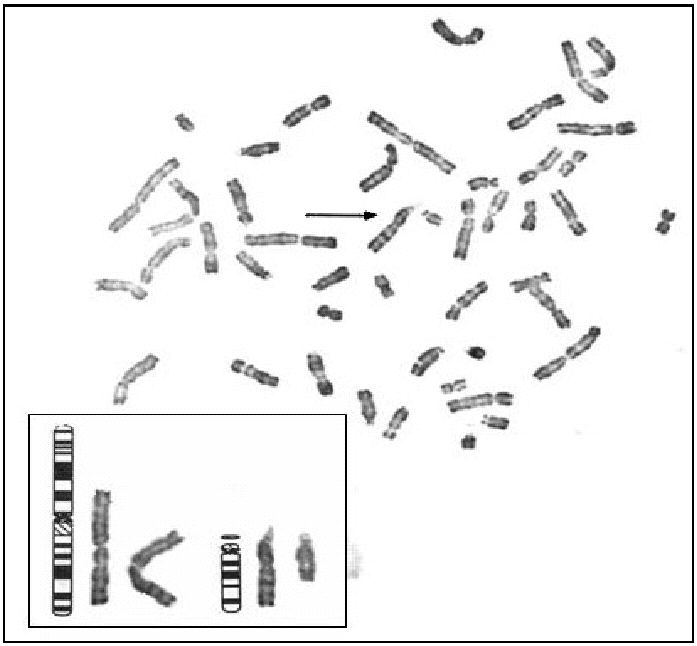
Tras el cultivo de las células procedentes del líquido amniótico, todas las metafases examinadas mediante bandas G pusieron de manifiesto, en los 2 cultivos realizados, la existencia de un cariotipo con un cromosoma derivativo 14. Realizado un estudio citogenético a los progenitores, el padre era portador de una translocación equilibrada t (1:14) (q25-q32) (fig. 2), por lo que puede concluirse que el cromosoma derivativo 14 era el resultado de una segregación adyacente I durante la meiosis paterna. Por tanto, el cariotipo corresponde a una trisomía parcial del cromosoma 1 que afecta al fragmento 1(q25-q44), y el diagnóstico fue citogenético: 46, XX, der(14) t (1;14) (q25-q32) pat (fig. 3).
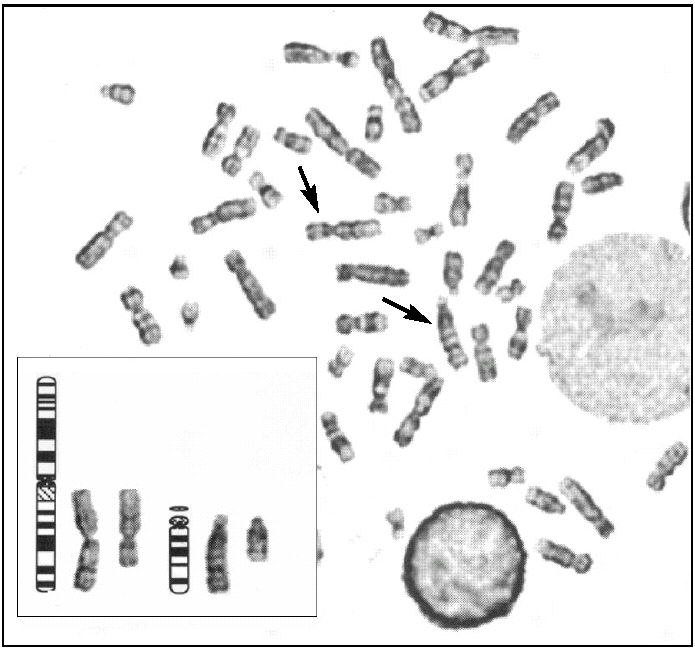
Figura 2.Cariotipo del cultivo de linfocitos paternos con translocación t(1;14) (q25;q32).
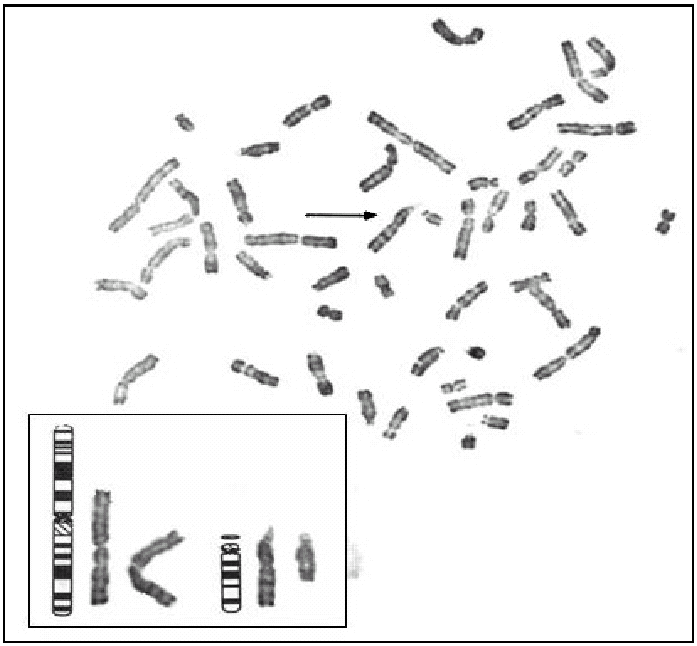
Figura 3.Cariotipo del cultivo de amniocitos, con trisomía parcial del cromosoma 1 por translocación paterna. 46,XX, t (1;14) (q25;q44:q32) pat.
Ante el diagnóstico citogenético y las malformaciones fetales existentes, la pareja decide interrumpir el embarazo.
En el estudio necrópsico se observa un feto hembra de 88,44 g de peso y 11,3 cm de longitud, con un cuadro polimalformativo consistente en:
-- Malformaciones externas: pabellones auriculares de implantación baja, higroma quístico de cuello, separación anómala entre los dedos 3 y 4 de ambas manos, y pies en mecedora con superposición del cuarto dedo sobre el tercero y quinto.
-- Malformaciones internas. Genitourinarias: riñón y uréter único de disposición central anterior con características de riñón poliquístico de Potter grado II-III, asociado a megavejiga. Hígado y vesícula biliar, con agenesia de vesícula biliar y fibrosis portal con proliferación de conductillos biliares. Hipoplasia tímica (fig. 4).
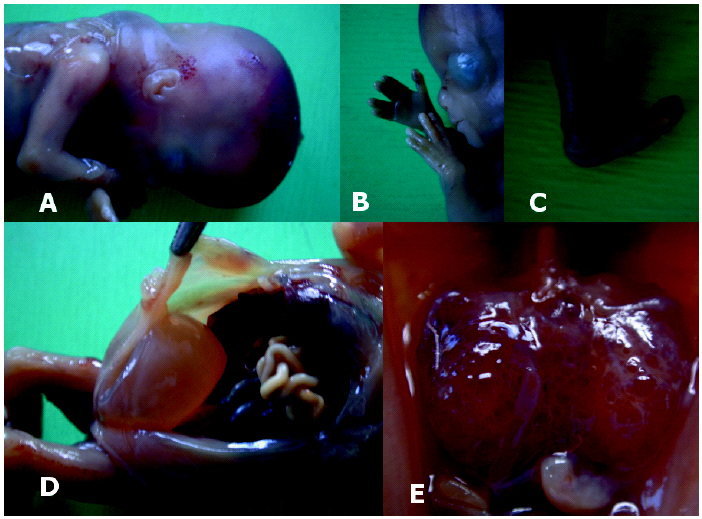
Figura 4.Hallazgos necrópsicos: A) Pabellones auriculares de implantación baja e higroma quístico. B) Separación anómala entre los dedos 3 y 4 de ambas manos. C) Pies en mecedora con superposición del cuarto dedo sobre el tercero y quinto. D) Megavejiga urinaria. E) Riñón único con características de riñón poliquístico con uréter único de disposición central anterior.
El resto de los hallazgos necrópsicos se corresponden con signos de shock intraútero agudo (congestión visceral generalizada y hemorragias parenquimatosas), así como los propios de la inmadurez fetal correspondientes a la edad gestacional. En el estudio de la unidad placentaria se observó una placenta de bajo peso para la edad gestacional con retraso en la maduración vellositaria y vellosidades grandes e irregulares con disminución del número de vasos, aumento de la celularidad y seudoinclusiones trofoblásticas.
DISCUSION
En la trisomía parcial del cromosoma 1 se han descrito diferentes puntos de rotura: 1q23, 1q25, 1q31, 1q32 y 1q427,8,9. Taysi y Sekhon10 sugieren que la duplicación parcial de los 2 tercios distales de 1q se caracteriza por malformaciones muy graves: fisura palatina, hipertelorismo, alteraciones renales, ausencia de vesícula biliar y muerte temprana11. Cuando la duplicación afecta sólo a un tercio distal de 1q se asocia a supervivencias más largas y a malformaciones medias y menos específicas, como orejas de implantación baja, micrognatia, anomalías cardíacas, dedos largos y testículos sin descender12, por lo que el fenotipo de este grupo de pacientes parece independiente del tamaño del segmento duplicado13.
En los casos descritos en la bibliografía con trisomía parcial por duplicación 1q25-qter14,15,16 se refiere un retraso del crecimiento psicomotor unido a riñones displásicos, atrofia cerebral, sistema biliar anormal, hipoplasias tímicas, malformaciones cardíacas y sistema biliar anormal, junto a micrognatia, orejas de implantación baja, paladar hendido e hipertelorismo. Las anomalías cardíacas y tímicas, así como las del sistema biliar y renal, son bastante características17.
En nuestro caso encontramos similitudes clínicas con los casos descritos en la bibliografía, como pabellones auriculares de implantación baja, dedos superpuestos, alteraciones renales, agenesia de vesícula biliar e hipoplasia tímica. Por tanto, creemos que el fenotipo puede ser atribuido a la trisomía parcial 1q25-qter, y que estos hallazgos pueden contribuir a la delimitación del síndrome de trisomía parcial 1q (q25-qter).
Correspondencia:
Dr. B. Correa Ceballos.
Avda. de Madrid, 3, 1.o 2.a B. 38007 Santa Cruz de Tenerife. España.
Correo electrónico: Baroncio1@yahoo.es
Fecha de recepción: 13/5/05.
Aceptado para su publicación: 11/10/05.