








TDP-43 es una proteína nuclear crítica para la sobrevida de las células nerviosas. En algunas proteinopatías aparecen formas patológicas con inclusiones citoplasmáticas vinculadas a afecciones degenerativas del SNC, tales como demencias frontotemporales (DFT) y/o enfermedad de las neuronas motoras.
ObjetivoUna revisión de la literatura neuroquímica, neuropatológica y clínico-neurológica acerca de TDP-43, respecto de su participación en algunas demencias degenerativas primarias, elaborando hipótesis acerca de la perturbación de funciones que podrían estar afectadas.
Desarrollo(1) Se describe la estructura de la proteína TDP-43 y se comentan sus funciones normales respecto de a) la regulación de la expresión de genes; b) control de la síntesis y metabolismo de los ARNs; c) su actividad en las sinapsis y en las respuestas de las células nerviosas al entorno; d) sus funciones tróficas; e) su función respecto del transporte de moléculas por los endosomas; f) su intervención en los procesos de ubiquitinación y proteólisis junto a autofagosomas y lisosomas; g) formar parte de los gránulos de stress que protegen a los ARNs ante condiciones dañinas; (2) se comenta la aparición de TDP-43 patológicas, mencionando infrecuentes mutaciones genéticas a nivel del gen TARDBP que la codifica y posibles alteraciones del plegamiento de la proteína; 3) se presentan variantes neuropatológicas de las DFT y genes asociados a ellas; 4) se comenta la presencia de inclusiones TDP-43 en el espectro de las enfermedades neuropsiquiátricas más frecuentes; 5) su presencia en el LCR; y 6) en diversos cuadros demenciales fuera del espectro de las DFT.
ConclusionesLas mutaciones del gen TARDBP son infrecuentes y no explican la aparición de las inclusiones citoplasmáticas de TDP-43 en la mayoría de los casos en que aparecen, incluso en casos no familiares. Los efectos neurodegenerativos de su disfunción obedecen a un complejo de factores, pero la vía final común es la conformación de agregados citoplasmáticos que pueden expandirse de un modo parecido a como los priones lo hacen. El goal standard es generar biomarcadores que puedan detectar a tiempo la presencia de estos agregados para usar fármacos que detengan su formación impidiendo la fosforilación y el clivaje de la proteína TDP-43 al tiempo de su depósito, anticipándose a la instalación del deterioro cognitivo. Tales biomarcadores pueden estar presentes entre los más de 28 anticuerpos contra TDP-43 patológicas que se hallan en estudio en el LCR en este momento. Todos estos conceptos valen para el espectro DFT-ENM; fuera de ese campo, la presencia de TDP-43 suele asociarse con esclerosis hipocampal (EH).
TDP-43 is a nuclear protein critical to nerve cells survival. In some proteinopathies pathological forms appear with cytoplasmic inclusions linked to degenerative CNS conditions, such as Frontotemporal Dementias (FTDs) and/or Motor Neurone Disease (MND).
ObjectiveA review of the neurochemical, neuropathological and neurological literature on TDP-43, regarding its participation in some primary degenerative dementias, developing hypotheses about functional disturbances.
Development(1) Description on TDP-43 structure and discussion about its functions regarding: a) regulation of gene expression, b) control of RNA's synthesis and metabolism, c) normal activity in synapses and in nerve cells responses to the environment, d) trophic functions, e) regulation of number and functions of endosomes, f) intervention in ubiquitination and proteolitic processes related also with autophagosomes and lysosomes, g) formation of stress granules protecting RNA from toxic conditions; (2) comments on the appearance of pathological TDP-43 are linked with infrequent genetic mutations at the level of the TARDBP gene that encodes TDP-43 and possibilities of protein misfolding; (3) presentation of neuropathological variants of FTDs and associated genes; (4) TDP-43 inclusions in the spectrum of the most common neuropsychiatric diseases and; (5) TDP-43 presence in the CSF and (6) TDP-43 in Dementias other than FTD.
ConclusionsMutations on the TARDBP gene are rare and do not explain the occurrence of cytoplasmic inclusions of TDP-43 in most cases where they occur, even in non-family cases. The neurodegenerative effects of their dysfunction are due to a complex of factors, but the common final pathway is the formation of cytoplasmic aggregates that can expand in a similar way to how prions do. The goal standard is to generate biomarkers that can detect in time the presence of these aggregates in order to use drugs that could stop their formation preventing the phosphorylation and clivation of the protein TDP-43 at the time of their deposit, anticipating the onset of cognitive decline. Such biomarkers may be present among more than 28 pathological TDP-43 antibodies currently under study in the CSF. All these concepts are suitable for the FTD-MND spectrum; outside that field, the presence of TDP-43 is frequently associated with hippocampal sclerosis (HS).
TDP-43 (proteína de respuesta transactiva conjugada a ADN de 43kDa o TAR DNA binding protein 43kd) pertenece a una familia de ribonucleoproteínas nucleares heterogéneas (hnRNP) localizada preferentemente en el núcleo de células nerviosas y gliales.
Está presente en todos los eucariotes, en tres isoformas, siendo crítica para la sobrevida de las células nerviosas. Aparecen formas patológicas que se manifiestan con inclusiones citoplasmáticas y nucleares neuronales y gliales en algunas proteinopatías ligadas a enfermedades degenerativas del SNC. En estos casos hay una depleción de la proteína en el núcleo debido a que se desvía hacia el citoplasma. La pérdida de la función de TDP-43 conduce a degeneración DFT-ENM (demencia frontotemporal-enfermedad de neurona motora), pero los mecanismos moleculares a través de los cuales esto ocurre son insuficientemente comprendidos.
La fracción nuclear de TDP-43 se une a muchos ARN vitales para la sobrevida y función de las neuronas, y controla el tráfico dendrítico de endosomas que reciclan1. La fracción citoplasmática es vital para el transporte de gránulos de ARNm.
El gen TARDBP está localizado en el cromosoma 1p36.22, donde 5 exones codifican TDP-43. Algunas mutaciones en este gen se vinculan a afecciones como la demencia frontotemporal (DFT-TDP) y la enfermedad de neurona motora (ENM, 10% de formas familiares)2,3, aunque hay además otros genes (MAPT, GRN, VCP, C9orf72, CHMP2B, FUS, etc.) implicados en estas enfermedades. La aparición de inclusiones patológicas de TDP-43 no queda limitada a las formas familiares4–8.
Ha sido recientemente demostrado que TDP-43 es una proteína que normalmente ayuda a regular la expresión de genes en el cerebro y otros tejidos9. Estudios previos encontraron que un plegamiento erróneo de TDP-43 tiene un papel causal en casos de esclerosis lateral amiotrófica de origen familiar y degeneración lobular frontotemporal ubiquitina-positivos tau-negativos10,11. La proteína TDP-43 mal plegada es común en adultos mayores. Aproximadamente el 25% de las personas mayores de 85 años tiene suficiente proteína TDP-43 mal plegada que podría estar afectando su memoria y/u otras funciones cognitivas. Borroni et al. (2009) describen una mutación TARDBP vinculada a un caso de DFTvc (variante comportamental). Una mujer septuagenaria con manifestaciones de apatía y disfunción ejecutiva fue diagnosticada como DFTvc. Extensas investigaciones electrofisiológicas excluyeron ENM. Se identificó en ella una mutación genética en TARDBP por sustitución de arginina por serina en posición 267 (N267S), mutación que ya había sido observada en pacientes con ENM. Sílico análisis demostraron que esta sustitución generaba una fosforilación «de novo». Los autores sugieren que estas mutaciones en TARDBP pueden ser patogenéticas para DFTvc sin ENM, y que el screening de TARDBP debe ser considerado mandatorio en estos casos12.
TDP-43 es una proteína nuclear de 414 aminoácidos que se encuentra muy activa durante el desarrollo embrionario, ligada al ciclo celular neuronal y al desarrollo neural. Pesa 44,74kd. TDP-43 está prominentemente expresada en hipocampo, células de Purkinje, glomérulo del bulbo olfatorio, y astas anteriores, sustancia blanca y astas posteriores de la médula espinal13.
Posee dos sitios de reconocimiento del ARN, llamados motivos RRM1 y RRM2, un terminal C-carboxílico rico en glicina (GRR), un sitio de autorregulación con un servomecanismo negativo cuyo déficit puede conducir a patología, dos sitios que regulan los pasajes entre núcleo y citoplasma llamados NLS (nuclear localization signal) y NES (nuclear export signal), y un N-terminal vinculado al binding de ADN y a la formación de homodímeros (fig. 1).
 RRM1. ARN binding, ARN splicing, autorregulación, gránulos de stress, conversión patológica y homodimerización. b) RRM2. ARN binding, formación de agregados, truncados y homodimerización. c) Región autorreguladora 321-366. ARN binding y splicing, junto a C-terminal. d) GRR (C terminal). Mutaciones TARDBP, interacciones proteína-proteína, autorregulación con «feed back» negativo, procesamiento y localización del ARN, dominio «priónico» y agregación. e) NLS. Controla la migración entre núcleo y citoplasma y la formación de agregados en el citoplasma. f) NES. Controla la migración citoplasma-núcleo y la formación de agregados en el núcleo. g) N-terminal. Plegamiento y homodimerización, splicing, agregación y ADN binding. Un cierto tráfico de TDP-43 entre el núcleo y el citoplasma es normal, pero predominando en el núcleo. Cuando se invierte la proporción, es patológico. TDP-43 es un regulador global de la expresión génica. Forma complejos con ribonucleoproteínas que regulan transcripción de genes, splicing y splicing alternativo, estabilidad, transporte y traducción del ARN, e influyen en la maduración de los microARN. Modificada de Janssens y van Broeckhoven4.")
Estructura, funciones y disfunciones en TDP-43: a) RRM1. ARN binding, ARN splicing, autorregulación, gránulos de stress, conversión patológica y homodimerización. b) RRM2. ARN binding, formación de agregados, truncados y homodimerización. c) Región autorreguladora 321-366. ARN binding y splicing, junto a C-terminal. d) GRR (C terminal). Mutaciones TARDBP, interacciones proteína-proteína, autorregulación con «feed back» negativo, procesamiento y localización del ARN, dominio «priónico» y agregación. e) NLS. Controla la migración entre núcleo y citoplasma y la formación de agregados en el citoplasma. f) NES. Controla la migración citoplasma-núcleo y la formación de agregados en el núcleo. g) N-terminal. Plegamiento y homodimerización, splicing, agregación y ADN binding. Un cierto tráfico de TDP-43 entre el núcleo y el citoplasma es normal, pero predominando en el núcleo. Cuando se invierte la proporción, es patológico. TDP-43 es un regulador global de la expresión génica. Forma complejos con ribonucleoproteínas que regulan transcripción de genes, splicing y splicing alternativo, estabilidad, transporte y traducción del ARN, e influyen en la maduración de los microARN. Modificada de Janssens y van Broeckhoven4.
A través de autointeracción en el citoplasma se forman homodímeros en RRM2, que aparecen en las inclusiones en las proteinopatías TDP-4314,15. TDP-43 forma complejos con varias ribonucleoproteínas. Se une a miles de ARN, preferentemente a intrones 3 y 5 de regiones sin traducción del ARN (UTRs). Actúa sobre ARN no codificantes y sobre ARNm presplicing (preensamblaje) regulando miles de transcripciones. Transcripciones para el desarrollo neural desde Notch1, MapT, PSD-95 y Dyrk1a, y para la guía de axones a partir de Ntn2, Sema3f, 4d, 6b y Ephb1-316.
En el núcleo aparece en fibrillas pericromatina que inciden en el splicing (ensamblaje). Y en «manchitas nucleares» donde se concentran los procesos postranscripcionales del pre-ARNm, formando sus propios corpúsculos: los «cuerpos TDP-43»17.
La proteína FUS/TLS (fused in sarcoma/translocated in liposarcoma) tiene la misma estructura RRM-GRR. Mutaciones en genes FUS pueden provocar ENM y DFT-U sin inclusiones de TDP-4318, pudiendo aparecer acumulada en agregados en DFT-FUS.
Funciones de TDP-43- 1)
Los residuos 321-366 son críticos para la interacción con hnRNPA2 formándose un complejo TDP-43/hnRNP que regula el splicing19.
- 2)
Durante el desarrollo embrionario, orienta el crecimiento de dendritas y axones hacia sus blancos11. Colocaliza con PSD-95 en la postsinapsis. Cuando el cerebro madura decae el nivel de TDP-43 mientras que PSD-95 funciona como marcador de maduración sináptica20.
- 3)
Incide en la adecuación de respuestas de las células nerviosas a su entorno, homeostáticas, inmunológicas y de reparación de tejidos, influyendo en la señalización de las mismas21, transportando ARNm a las sinapsis.
- 4)
TDP-43 en motoneuronas regula la morfología y el desarrollo de espinas dendríticas y plasticidad neuronal. Si falta TDP-43 hay bloqueo de señales tróficas y de transporte y reciclado vesicular, disminuyendo la motilidad y el número de endosomas dendríticos RAB-11 (la hiperexpresión de TDP-43 tiene efectos opuestos) con depleción de receptores ErbB4 (neurorregulina 1R). Hay concomitantemente up-regulation del componente ESCRT-VPS4B, porque TDP-43 reprime la transcripción del gen VSP4B. Antagonizando a VSP4B se normalizan los endosomas, al igual que con la hiperexpresión de ErbB41. Muchas proteínas con reducida expresión superficial están implicadas en el crecimiento dendrítico (ErbB4, FGFR1, EPhB2) o axonal (Robo1, Unc5c/d, EphB2, TrkB)16. TDP-43 controlaría la expresión de esos receptores necesarios para el crecimiento de los axones y la sobrevida de las neuronas. La remoción de los agregados TDP-43 y la restauración de su expresión nuclear provocan una reinervación funcional en un modelo murino que abonaría la hipótesis de las funciones tróficas de TDP-4322.
- 5)
Se vincula a la potenciación de largo término (LTP)21.
- 6)
La alteración de la autofagia puede gatillar DFT-TDP. La disfunción lisosomal agrava el cuadro. Mutaciones disruptivas del tráfico endocítico y de la fusión de autofagosomas con lisosomas favorecen el desarrollo de DFT-UPS; por ejemplo: CHMP2B (no aparecen inclusiones TDP-43)23. TDP-43 regula la localización lisosomal del factor de transcripción EB (TFEB) a través del complejo raptor/mTORC1. El déficit de TDP-43 provoca: a) traslocación nuclear de TFEB, con aumento de la expresión de genes de autofagosomas y síntesis de lisosomas; y b) disminución de la fusión de autofagosomas con lisosomas por down-regulation de α dinactina 1, con acumulación de vesículas autofágicas inmaduras. En un modelo de Drosofila con depleción de TDP-43, la inhibición de mTORC1 agrava el fenotipo de la degeneración, en tanto que su activación lo mejora. Se concluye que el déficit de mTORC1 contribuye a provocar los efectos neurodegenerativos de TDP-4324. La perturbación de la autofagia puede conducir a la acumulación anormal de proteínas (proteinopatía). La acumulación de autofagosomas obra como un stressor, induciendo toxicidad por deterioro del sistema autofagosomas-lisosomas. Se plantean como estrategias terapéuticas: a) detener la acumulación de vesículas de autofagosomas, y b) restaurar la fusión de autofagosomas con lisosomas24.
- 7)
El fallo de TDP-43 conlleva menor transporte a través del núcleo y menor degradación de proteínas. El sistema proteolítico requiere chaperones moleculares asociados a UPR (respuesta a la acumulación de proteínas), UPS (ubiquitina-proteasoma) y/o autófagos lisosomales25. Si se inhibe dicho sistema aumenta la agregación de TDP-4326, con alteración de importinas, que regulan el tráfico de moléculas a través de la membrana nuclear. El receptor P62 contribuye a normalizar la situación activando el sistema proteolítico27.
Entre las funciones de TDP-43 se destacan: (i) regular la síntesis y el metabolismo de miles de ARN, (ii) reprimir la transcripción de algunos genes, (iii) operar el ensamblaje alternativo de algunos ARNm, (iv) transporte y estabilización de algunos ARN, (v) unirse a ARN no codificantes y a miARN, y (vi) influir el metabolismo del ARN durante la respuesta al stress térmico, mecánico o tóxico. Se une a los ARN a través de bases UG repetitivas en regiones intrónicas28. Además TDP-43 interviene en: a) salteo exónico del gen regulador de la conductancia transmembrana en la fibrosis quística29 y genes de apoliproteínas I y II30, b) inclusión exónica del gen para la sobrevida de la motoneurona31, c) estabilización de la proteína ARNm de bajo peso molecular de los neurofilamentos32, d) modulación de la expresión ciclinadependiente de kinasa 633 y microARN biogénesis34, e) transporte de ARNm y regulación en las sinapsis35, f) regulación del ciclo celular21, y g) apoptosis36. Cumple estas funciones uniéndose a ADN, ARN y/o proteínas16. Sus oligómeros pueden atravesar las sinapsis. Su terminal carboxílico, rico en glicina, se une a muchas ribonucleoproteínas que intervienen en la biogénesis de ARNm, controlando la síntesis de muchas otras4.
Los gránulos de stress (SG), producidos por stress oxidativo, contienen agregados insolubles de TDP-43 y ribonucleoproteínas, que reflejan intentos de traducción estancados. Protegerían a los ARNs ante condiciones dañinas. Son una expresión intermedia entre TDP-43 normal e inclusiones patológicas37.
TDP-43 patológicaProteína mal plegada con conformación de agregados proteicos10. Las TDP-43 patológicas presentan fosforilación, clivaje en C-terminal y agregación citoplasmática (fig. 2).
 Localización nuclear de TDP-43 normal. B) Localización patológica extranuclear.")
La forma patológica de TDP-43 se encuentra hiperfosforilada en los segmentos 379, 403, 404, 409 y 410 y truncada en el terminal N38,39. Sufre clivaje en fragmentos de menor peso molecular, de 35 y 25kd, que no contienen el terminal N, disminuyendo su solubilidad y alterándose los procesos de ubiquitinación40 en las regiones afectadas.
La TDP-43 patológica forma agregados que se van expandiendo de un modo parecido a los priones41. Fragmentos fosforilados C-terminal han sido detectados en la corteza cerebral de pacientes con DFT-U y ENM, siendo más abundantes que la proteína fosforilada completa42. Pueden aparecer en relación con mutaciones de TARDB-P o de otros genes, o en casos esporádicos no familiares. Otros genes relacionados con el complejo ENM-DFT-U (ubiquitina) son: GRN43–47, ANG48, hnRNPA1, hnRNPA2/B149, C9orf7250, ATXN251, VCP52, UBQLN253, OPTN54 y SQSTM155,56. Se sospecha que hay alteración en el procesamiento de ARN y en la degradación de proteínas.
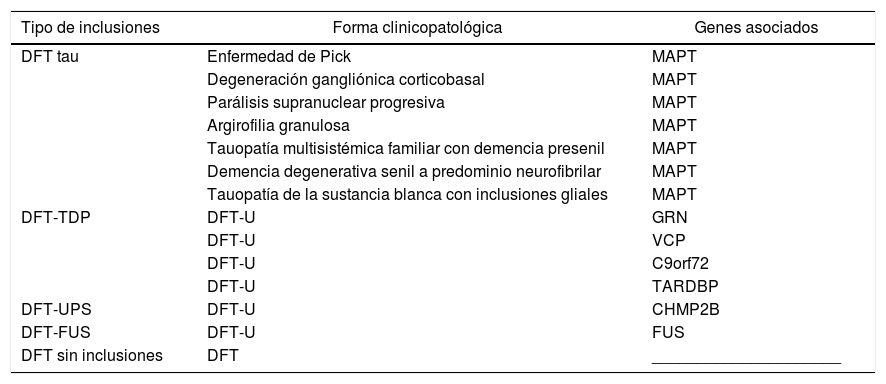
Genética molecular y neuropatología en demencia frontotemporalEstas TDP-43 patológicas dan origen a TDP-proteinopatías, una clase de desórdenes neurodegenerativos, que incluyen también a otras clases como las tauopatías (tabla 1).
Variantes neuropatológicas en demencia frontotemporal y genes asociados
| Tipo de inclusiones | Forma clinicopatológica | Genes asociados |
|---|---|---|
| DFT tau | Enfermedad de Pick | MAPT |
| Degeneración gangliónica corticobasal | MAPT | |
| Parálisis supranuclear progresiva | MAPT | |
| Argirofilia granulosa | MAPT | |
| Tauopatía multisistémica familiar con demencia presenil | MAPT | |
| Demencia degenerativa senil a predominio neurofibrilar | MAPT | |
| Tauopatía de la sustancia blanca con inclusiones gliales | MAPT | |
| DFT-TDP | DFT-U | GRN |
| DFT-U | VCP | |
| DFT-U | C9orf72 | |
| DFT-U | TARDBP | |
| DFT-UPS | DFT-U | CHMP2B |
| DFT-FUS | DFT-U | FUS |
| DFT sin inclusiones | DFT | _____________________ |
C9orf72: complejo con subunidad SMCR8 locus genético en cromosoma 9p; CHMP2B: gen de proteína cargada con cuerpo multivesicular; DFT: demencias frontotemporales; FUS: gen de proteína fused in sarcoma traslocada a liposarcoma (FUS/TLS)102; GRN: gen de la progranulina; MAPT: gen de proteína tau asociada a microtúbulos; TARDBP: gen de la proteína de respuesta transactiva conjugada a DNA de 43kd; TDP: proteína TDP-43; U: ubiquitina; UPS: sistema ubiquitina-proteasoma; VCP: gen de proteína valosinada.
Recientes estudios han mostrado la acumulación de TDP-43 en las mitocondrias de neuronas de pacientes con ENM y DFT, revelando que las mitocondrias pueden ser un mediador crítico de la neurotoxicidad57.
- 1)
Tau+ubi–: p.ej., mutación en gen de MAP-T en el cromosoma 17. Se presenta clínicamente como DFT con parkinsonismo58.
- 2)
TDP-43 asociada a mutaciones del gen GRN en el cromosoma 1746,47: la insuficiencia de PGR se liga a la DFT-U con patología TDP-4359. La enfermedad comienza alrededor de los 60 años y no suele asociarse a ENM60. La pérdida de función del gen de la PGR en un 50% es causa principal de DFT-U familiar (25% de los casos de DFT-U), por haploinsuficiencia61. La disminución de la expresión del gen GRN in vitro ha sido asociada con aumento del clivaje y acumulación patológica de TDP-4344–46. El espectro de presentaciones clínicas en estos casos es muy amplio. La PGR tiene efectos como factor neurotrófico y acciones antiinflamatorias débiles, pero sometida a la acción de proteasas, origina granulinas de acciones fuertemente proinflamatorias62. PGR actúa en: desarrollo, reparación de heridas, inflamación, cascadas de activación de señales y control de la progresión del ciclo celular y de la motilidad. Un cut-off plasmático de GRN 110,9ng/ml distingue a los portadores de la mutación con S=100 y E=92,863. Gliebus et al. (2010) encontraron una distribución muy asimétrica de las inclusiones TDP-43 en un caso con mutación PGR, más evidente en el giro parietal inferior y en los giros temporales superior e inferior, y orbitofrontal, a predominio izquierdo43. La misma mutación pudo causar afasia progresiva primaria (APP) en un miembro de la familia y DFTvc en otro64.
- 3)
La TDP-43 asociada a VCP en el cromosoma 965 está comúnmente acompañada de miositis por cuerpos de inclusión y enfermedad de Paget ósea66. La presentación clínica incluye DFTvc o APPvs (variante semántica) y comienza entre los 50 y 60 años67. Neuropatológicamente, este subgrupo de DFT-TDP se distingue por la combinación de inclusiones intranucleares TDP-43 positivas con neuritas distróficas con pocas inclusiones citoplasmáticas intraneuronales68,69.
- 4)
TDP-43 asociada a TARDBP en el cromosoma 170: más de 25 mutaciones del gen TARDBP han podido ser identificadas, la mayoría en el dominio GRR; afectando ensamblaje (splicing) y saltos (skipping)5.
- 5)
La TDP-43 está asociada a mutaciones C9orf72, con repetición de hexanucleótidos en cromosoma 950. Su ubicación citogenética está en el brazo corto p21.2. Provoca ENM familiar71.
- 6)
No hay TDP-43 en DFT con proteína FUS, en casos esporádicos no familiares. Comienza entre los 30 y 45 años provocando además de la DFT un síndrome akineto-rígido con atrofia cortical, de estriado e hipocampos. Se ha subdividido en tres grupos: a) DFT-FUS, b) NIFID (inclusiones de agregados de filamentos intermedios en la patología) y c) BIBD (aparecen cuerpos de inclusión basofílicos), que se distingue de los otros grupos porque afecta también a las motoneuronas72.
- 7)
Las mutaciones CHMP2B en cromosoma 3 son TDP-43 negativas. La atrofia en neuroimágenes es más difusa que la típica DFTvc33. La neuropatología es única porque las inclusiones citoplasmáticas ubiquitinadas no contienen tau, TDP-43 ni FUS (DFT-UPS, sistema ubiquitina-proteasoma)73 (fig. 3).
 Figura 3.
Figura 3.Neuropatología y genética en DFTvc. El espectro de las DFTvc se ha dividido en subtipos neuropatológicos según color y causas genéticas en letras negras fuera del círculo. DFT-TDP en púrpura, aproximadamente el 50% de casos. DFT-tau en naranja, aproximadamente 40%. DFT-FUS en rojo, menos de un 10% de casos. El resto, en verde, presenta DFT-UPS negativa para tau, TDP-43 y FUS. Mutaciones MAPT, C9ORF72 y GRN son las causas más comunes de las DFT genéticas. MAPT vinculadas a patología tau. C9ORF72 y GRN vinculadas a patología TDP-43, al igual que mutaciones en TARDBP y VCP. Mutaciones FUS causan patología FUS, mientras que las CHMP2B provocan FTD-UPS. Modificada de Roberson110.

DFT U+tau– (no siempre)5.
Miopatía con Paget óseo y DFT69.
Síndrome de Perry. Se afecta el gen DCTN1, que codifica a la dinactina 1. El síndrome de Perry es una enfermedad cerebral progresiva caracterizada por 4 signos principales: un patrón de movimientos anormales (parkinsonismo), cambios psiquiátricos, pérdida de peso e hipoventilación alveolar. Los signos de parkinsonismo incluyen bradicinesia, rigidez y temblores74.
SecundariamenteComplejo de la isla de Guam75,76, esclerosis hipocampal (EH)77, enfermedad de Alzheimer (EA)77,78 degeneración corticobasal78, enfermedad de Parkinson79, parálisis supranuclear progresiva80. También puede aparecer en agregados poliglutamínicos en la Corea de Huntington81.
MínimamenteEnfermedad de cuerpos de Lewy (LBD)82, enfermedad de Pick83, esclerosis lateral primaria84, DFT U+TDP43–85, atrofia multisistémica86, enfermedad priónica5, esquizofrenia87.
Puede haber colocalización TDP-43 con patología tau88 o con α sinucleína en raros casos89.
Localización de las inclusiones citoplasmáticasTDP-43 en líquido cefalorraquídeoLa TDP-43 puede aparecer truncada en el cerebro de pacientes con DFT-ENM, pero estas formas de 25 a 35kd no aparecen en el LCR, aunque en DFT-ENM, APPnf (afasia progresiva primaria no fluente) y APPvs pueden aparecer anticuerpos policlonales para terminal C de TDP-4391.
La inmunohistoquímica, la inmunofluorescencia, el inmunoblotting y el método enzyme-linked immunosorbent assays (ELISA) son técnicas de laboratorio idóneas para detectar la presencia de TDP-43.
La proteína TDP-43 es considerada un posible marcador para algunos subtipos de DFT. El objetivo a investigar es ¿qué anticuerpos son específicos para las isoformas patológicas de TDP-43, frecuentemente hiperfosforiladas y/o truncadas? A través de una revisión de la literatura, Goossens et al. (2015) encuentran 29 anticuerpos con propiedad de detectar TDP-43 patológicas92. Algunos de estos anticuerpos podrían funcionar como marcadores en biofluidos en proteinopatías caracterizadas por la presencia de inclusiones citoplasmáticas de TDP-43.
El objetivo de un biomarcador93 es: 1) contribuir a un diagnóstico preclínico, 2) diferenciar una enfermedad de otras, 3) predecir la progresión de la enfermedad, 4) contribuir a comprender su fisiopatología, 5) caracterizar imágenes específicas y 6) comprobar la efectividad de una terapéutica. Sus requisitos son: 1) estar validado por la histopatología, 2) tener una sensibilidad y especificidad mayor del 80%, 3) resultados reproducibles, 4) aplicación incruenta, y 5) aceptable relación costo-beneficio.
TDP-43 patológica – algunas consecuencias posiblesPuede haber aumento o disminución en el cumplimiento de sus funciones, predominando las hipótesis deficitarias94. Conllevaría:
- 1)
trastornos en la expresión genética y en la regulación de la actividad y el metabolismo de los ARN;
- 2)
disminución del número y actividad de endosomas dendríticos1;
- 3)
alteración de la regulación de los sistemas autofagosomas-lisosomas y proteasomas37;
- 4)
disminución del reciclado de receptores en la superficie celular, pudiendo alterarse señales tróficas1;
- 5)
eventualmente daño en la función mitocondrial95;
- 6)
afectación de la sobrevida de las neuronas motoras.
Las proteínas pueden sufrir modificaciones postraduccionales, como por ejemplo la ubiquitinación96, adición de una o varias moléculas de ubiquitina, un pequeño péptido, de manera covalente al blanco. Hay tres enzimas intervinientes: 1) E1 de activación, 2) E2 de conjugación, y 3) E3 que define la especificidad de la reacción, dirigiendo las E2 a las proteínas blanco97 (fig. 4).
. Ubiquitina es activada por E1, conjugada por E2 y ligada al sustrato por E3. Proteínas mal plegadas actúan como sustratos del UPS, reconocidos por chaperones moleculares y asociados con Ub ligasas que promueven la transferencia de E2-Ub y E3-Ub a dichos sustratos. Los sustratos son luego captados por el receptor UBA y el proteasoma 19S y progresivamente degradados a pequeños péptidos por el proteasoma 26S97.")
Degradación de las proteínas por el sistema UPS (ubiquitinación-proteólisis). Ubiquitina es activada por E1, conjugada por E2 y ligada al sustrato por E3. Proteínas mal plegadas actúan como sustratos del UPS, reconocidos por chaperones moleculares y asociados con Ub ligasas que promueven la transferencia de E2-Ub y E3-Ub a dichos sustratos. Los sustratos son luego captados por el receptor UBA y el proteasoma 19S y progresivamente degradados a pequeños péptidos por el proteasoma 26S97.
La ubiquitinación de proteínas lleva a cabo gran cantidad de funciones por medio del marcaje con ubiquitinas de proteínas celulares con consecuencias muy diversas, desde cambios de localización dentro de la célula hasta la activación, inactivación, o degradación de las mismas.
La ubiquitina es regulatoria para remover situaciones de defecto o exceso en el funcionamiento de MAPT, PGR y/o TDP-43. Formas autosómicas dominantes de DFT llegan hasta el 27%, el 11% corresponden a MAPT, el 6% a PGR y el resto a otras causas. Los pacientes con mutaciones PGR tienen un promedio de edad de 62 años, contra 52 años los de MAP-T98.
TDP-43 en otros cuadros demencialesTDP-43 en afasia progresiva primariaHa sido descripta una disociación en la patología cortical entre la DFT-TDP, que predomina en las regiones temporal y ventral frontal y la DFT-tau cuya patología es dorsolateral frontal98. Giannini et al. (2019) reportan 8/9 pacientes con APPvs (variante semántica) con patología DFT-TDP y 10/13 con APPnf con patología DFT-tau, corroborando datos anteriores99,100.
Los fenotipos de APP/DFT en casos con patología de Alzheimer tienen lesiones bihemisféricas que no suelen asociarse con inclusiones TDP-43, las cuales son más frecuentes en la variante semántica99,100. Cuando aparecen están más ligadas a EH que al fenotipo clínico. Parecería que TDP-43 puede contribuir a su patogenia sólo en un limitado número de casos de APP77,101. En otros pueden aparecer inclusiones tau o FUS73,102.
La pérdida neuronal y la gliosis en la región subicular pero no en el hipocampo mismo son predictivas de encontrar el subtipo con inclusiones TDP-4377.
TDP-43 en esclerosis hipocampalEstudios autópsicos en la población mayor de 65 años, y particularmente en los mayores de 85, han identificado ∼ un 20% de pacientes con deterioro cognitivo amnésico en los cuales el único dato patológico es la EH. Pero en otros pacientes la EH puede ser comórbida con DFT, EA o LBD. La EH que acompaña al envejecimiento suele presentar inclusiones TDP-43, incluso fuera del hipocampo103. En la DFT puede tratarse de un subtipo amnésico de DFT-U104.
Ha sido detectada inmunorreactividad para TDP-43 en el 71% de los casos con EH que acompañan a la EA. La doble tinción de los casos con EA para detectar TDP-43 y fosfo-tau mostró que las inclusiones TDP-43 no se hallan en las lesiones degenerativas neurofibrilares. Se asocian a gránulos y filamentos citosólicos, pero casi nunca a filamentos tau. Los Western blots de estos casos revelaron una banda que migró a un peso molecular más pesado, lo que no ocurrió en casos de EA sin inmunorreactividad para TDP-43105.
Según López et al. (2016), la EH y su correlato de inclusiones TDP-43 aparecen en la EA en función de mayor edad, larga duración de la enfermedad y presencia de cuerpos de Lewy106.
TDP-43 en degeneración gangliónica corticobasalSe halló patología TDP-43, predominantemente glial, en el 15,4% de 39 casos de degeneración corticobasal78. En estos casos la TDP-43 patológica se presentó en las células granulares de la fascia dentada y en la corteza entorrinal, resultando reactivos los oligodendrocitos en la subtancia blanca con presencia de corpúsculos similares a los cuerpos de Cajal. Por inmunofluorescencia se observó superposición parcial de la reactividad para ambas proteínas, tau y TDP-43. A nivel de los oligodendrocitos las inclusiones citoplasmáticas se marcan con alguna de las dos, pero no con ambas superpuestas. Hay también inclusiones nucleares en el 50% de los casos positivos. La TDP-43 patológica es similar a la hallada en DFT-TDP: truncada, hiperfosforilada y ubiquitinada. No hubo diferencias clínicas en cuanto a la duración de la enfermedad o la causa de muerte entre los grupos con o sin inclusiones TDP-4378.
TDP-43 en parálisis supranuclear progresivaEn la parálisis supranuclear progresiva hay datos conflictivos. Si hay inclusiones TDP-43 aparecen en amígdala y giro dentado. Cuando aparecen, concomitantemente aparecen más depósitos de tau. La correlación es más significativa en la circunvolución occipitotemporal. La EH está asociada a los casos con inclusiones de TDP-43. La demencia es más frecuente cuando aparece TDP-43 en la patología y cuando hay EH. TDP-43 y tau frecuentemente colocalizan en la amígdala pero no en el giro dentado80.
TDP-43 en enfermedad de AlzheimerLas lesiones características de la EA no suelen presentar inclusiones TDP-43, las cuales aparecen sólo en un 25% de los casos; y asociadas linealmente con EH107. Están en un 75% restringidas al córtex entorrinal y al giro dentado; el otro 25% en lesiones corticales frontotemporales en las capas superiores también, asemejándose a una DFT-U grupo 3 de Sampathu78,101. Se asocian a mayor patología y severidad de la expresión clínica y a una duración prolongada, pero no a una determinada presentación clínica, no siendo un marcador específico sino un signo que acompaña procesos terminales.
Las inclusiones TDP-43 pueden coexistir con patología tau, la cual puede antecederle, incluso en las mismas neuronas. Una severa pérdida de neuronas en hipocampo y subiculum (EH) se encontró en el 11,5% de los casos con diagnóstico patológico de EA asociada a patología TDP-43; en el 71,4% de esos casos, especialmente en las mismas regiones CA1 y subiculum78,105,108. La EH se presentaba en igual proporción en los casos con patología limitada a la región límbica respecto de aquellos con patología más difusa, siendo el porcentaje de presencia de TDP-43 similar en los casos con presentación clínica amnésica o conductual78.
Se concluye que los casos de APP con patología de tipo Alzheimer no suelen asociarse con proteinopatía TDP-43, a menos que haya esclerosis hipocámpica (Bigio et al., 2010)77.
Presencia de TDP-43 en el envejecimiento y en otras demenciasNakashima-Yasuda et al., 200782.
- •
LBD+Alzheimer=25/80 (31,3%).
- •
Parkinson con demencia=4/21 (19%).
- •
Parkinson=5/69 (7,2%).
La sustitución p.N267S en el gen TARDBP ha sido implicada en ENM y DFT, pero se reporta también enfermedad de Parkinson79. Hay más de 15 casos con mutaciones de ese gen y Parkinson hasta la fecha109.
- •
Envejecimiento normal=1/33 (3%).
- •
LBD=0/10 (0%).
Las mutaciones del gen TARDBP son infrecuentes y no explican la aparición de las inclusiones citoplasmáticas de TDP-43 en la mayoría de los casos en que aparecen, incluso en casos no familiares.
Los efectos neurodegenerativos de su disfunción obedecen a un complejo de factores, pero la vía final común es la conformación de agregados citoplasmáticos que pueden expandirse de un modo parecido a como los priones lo hacen.
El goal standard es generar biomarcadores que puedan detectar a tiempo la presencia de estos agregados para usar fármacos que detengan su formación impidiendo la fosforilación y el clivaje de la proteína TDP-43 al tiempo de su depósito, anticipándose a la instalación del deterioro cognitivo.
Tales biomarcadores pueden estar presentes entre los más de 28 anticuerpos contra TDP-43 patológicas que se hallan en estudio en el LCR en este momento.
Todos estos conceptos valen para el espectro DFT-ENM; fuera de ese campo, la presencia de TDP-43 suele asociarse con EH.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
