








Describir características clínicas de niños con atrofia muscular espinal (AME) seguidos en un hospital pediátrico durante 2021.
Pacientes y métodosEstudio observacional, transversal y retrospectivo.
ResultadosDe 74 pacientes incluidos, 41,9% fueron mujeres, con una mediana de edad de 96 (rango intercuartílico [IQR] 60-135) meses. Pacientes AME tipo 1 n=16 (24,6%); dos n=41 (55,4%); y tres n=17 (23%). La mediana de edad de diagnóstico para AME 1 fue: seis meses (IQR 4,2-7,7); dos: 16 meses (IQR 12-24) y tres: 36 (IQR 24,5-48) meses. Se describió el estado motor actual: 23 (31,1%) sujetos perdieron logros motores, cuatro (5,4%) ganaron habilidades motoras y 47 (63,5%) mantuvieron el nivel funcional. Sobre el acceso a estándares de cuidado, 55 (74,3%) realizaban tratamiento kinésico motor y 67 (90,5%) tenían equipamiento adecuado. Un total de 58 niños (78,3%) tenían escoliosis. Sobre el perfil respiratorio, 65 (87%) sujetos realizaban técnicas kinésicas y 31 (42,4%) requerían soporte ventilatorio. Un total de 14 pacientes (18,9%) requirieron internación por intercurrencias respiratorias. Sobre el tratamiento farmacológico, 40 participantes (54%) recibieron nusinersen.
ConclusiónDe los 74 pacientes, la mayoría fueron AME 2, de género masculino, provenientes del Gran Buenos Aires y estaban escolarizados. Más de la mitad mantuvo el logro motor más alto para su clasificación y fenotipo. La ventilación no invasiva (VNI) fue el método predominante de ventilación. La incidencia de internaciones por causas respiratorias fue baja. La mayoría accedió al tratamiento médico y equipamiento necesario.
To describe the clinical characteristics of children with spinal muscular atrophy (SMA) followed at a Paediatric Hospital during 2021.
Patients and methodsObservational, cross-sectional, retrospective study.
ResultsOf 74 patients included 41.9% were female, median age 96 (RIQ 60 - 135) months. Patients SMA type 1 n=16 (24.6%); 2 n=41 (55.4%); 3 n=17 (23%). Median age at diagnosis for SMA 1: 6 (RIQ 4.2 - 7.7) months; 2: 16 (RIQ 12 - 24) months and 3: 36 (RIQ 24.5 - 48) months. Current motor status was described: 23 (31.1%) subjects lost motor achievements, 4 (5.4%) gained motor skills and 47 (63.5%) maintained functional level. Regarding access to standards of care, 55 (74.3%) had access to physical therapy and 67 (90.5%) had adequate equipment. 58 children (78.3%) had scoliosis. Regarding the respiratory profile, 65 (87%) subjects were performing airway clearance techniques; 31 (42.4%) required ventilatory support. 14 (18.9%) patients required hospitalization for respiratory intercurrences. Regarding pharmacological treatment, 40 (54%) participants received nusinersen.
ConclusionOf the 74 patients, the majority were SMA 2, male, from Buenos Aires area and were enrolled in school. More than half maintained the highest motor achievement for their classification and phenotype. Noninvasive ventilation was the predominant method of ventilation. The incidence of hospitalizations for respiratory causes was low. Most had access to the necessary medical treatment and equipment.
La atrofia muscular espinal (AME) es una enfermedad neuromuscular que se caracteriza por la degeneración de las neuronas motoras en el asta anterior de la médula espinal1,2. Constituye la segunda causa más frecuente de enfermedad autosómica recesiva después de la fibrosis quística2,3. Los humanos tienen dos copias de genes casi idénticas, Survival Motor Neuron (SMN) 1 y SMN2. La deleción homocigótica del gen SMN1 causa la enfermedad2,4,5, y el número de copias del gen SMS2 modula el fenotipo, condicionando la gravedad.
La incidencia es de aproximadamente uno en 11.000 nacidos vivos1,4 sin conocerse cifras exactas en nuestro país. En cuanto a la prevalencia por subtipo, esta es variable5. La bibliografía refiere que, en países como Colombia y Cuba, el subtipo más común fue el 23,6, mientras que, en España fue el tipo 17. Hay una ligera predominancia del género masculino, con una relación 1,6:16–8. Existen cuatro subtipos de la enfermedad definidos según la edad de inicio y los logros motores alcanzados9. Varían desde la manifestación muy severa in utero a una forma leve de inicio en la edad adulta. La edad de inicio de los síntomas es variable con relación al fenotipo3,7,10,11. A pesar de que la bibliografía remarca la importancia de un diagnóstico temprano algunos trabajos reportan hasta 96 meses de diferencia entre la sospecha y la confirmación diagnóstica para los AME 33.
En la actualidad, la implementación de consensos sobre el manejo interdisciplinario y las nuevas terapias farmacológicas disponibles son elementos que pueden modificar la historia natural de la enfermedad. Según nuestro conocimiento, en Argentina no existen estudios epidemiológicos que describan esta población. Consideramos necesario establecer el perfil clínico de estos pacientes en nuestro contexto para sentar bases para futuros trabajos y relevar el acceso a los recursos en salud disponibles y necesarios para esta población.
El objetivo de este estudio es describir las características de la población de pacientes con AME tipo 1, 2 y 3 en seguimiento en un hospital público pediátrico en Argentina durante el año 2021.
Pacientes y métodosDiseñoEstudio descriptivo, observacional y transversal de carácter retrospectivo.
ParticipantesPacientes pediátricos diagnosticados con AME tipo 1, 2, y 3 en seguimiento en un hospital de pediatría ubicado en la Ciudad Autónoma de Buenos Aires. Se incluyeron niños de ambos sexos, con diagnóstico por estudio genético molecular de AME menores de 18 años, que tuvieran al menos una consulta en el año 2021 y con la historia clínica completa, es decir con al menos el 90% de las variables analizadas. Se excluyeron pacientes que tuvieran alguna otra patología asociada no relacionada.
Recolección de datosLa información fue recolectada a partir de la historia clínica informatizada y luego cargada y codificada en una base de datos realizada en una planilla Excel. Este trabajo fue evaluado y aceptado por el Comité Revisor y de Ética de Investigación del Hospital para su publicación.
VariablesSe recolectaron variables para caracterizar el perfil demográfico, motor y respiratorio de la población y otras relacionadas al diagnóstico. Las variables relacionadas a la edad se expresaron en meses. El perfil motor se definió como el hito del desarrollo más alto alcanzado de manera independiente y sin uso de equipamiento, considerándose el empeoramiento como la pérdida de un hito motor alcanzado y la mejoría como alcanzar un logro motor no esperable según su fenotipo. Dentro del perfil respiratorio se consideró ventilación parcial, aquella que se realiza durante menos de 16 horas al día.
Análisis estadísticoLas variables categóricas se reportaron como número de presentación y porcentaje. Las variables continuas que asumieron una distribución normal se reportaron como media y desviación estándar (DE). De lo contrario, se utilizó la mediana y rango intercuartílico (IQR). Para determinar la distribución de la muestra se utilizó el test de Kolmogórov-Smirnov. Para el análisis de los datos se utilizó el software Statistical Package for the Social Sciences (SPSS) para Windows® (versión 25.0, IBM [International Business Machines] Corp. © Copyright IBM Corporation y sus licenciatarios 1989, 2019).
ResultadosCaracterísticas de la muestraDurante 2021, fueron evaluados 77 pacientes con AME, tres fueron excluidos del análisis por presentar patologías concomitantes y no relacionadas. En la figura 1 se presenta el diagrama de flujo de los participantes junto a las causas de exclusión.

A continuación, se exponen las causas de exclusión.
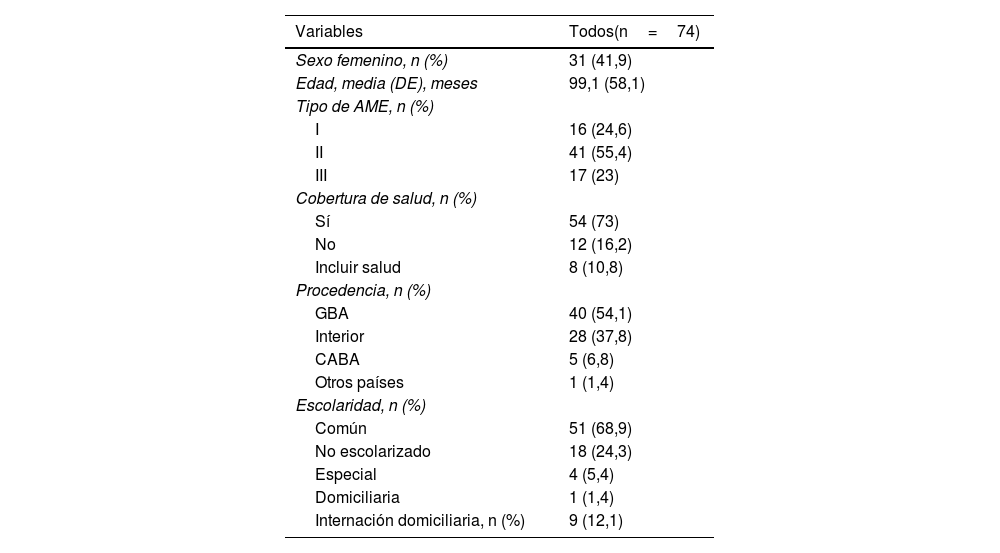
Características demográficas y clínicasDel total de pacientes incluidos, 31 (41,9%) fueron mujeres, con una mediana de edad de 96 meses (IQR 60-135). Con relación a los antecedentes, tres pacientes no contaron con dicha información, mientras que 18 (24,3%) refirieron poseer algún familiar con diagnóstico de AME. En la tabla 1 se presentan las variables sociodemográficas.
Características sociodemográficas de la muestra
| Variables | Todos(n=74) |
|---|---|
| Sexo femenino, n (%) | 31 (41,9) |
| Edad, media (DE), meses | 99,1 (58,1) |
| Tipo de AME, n (%) | |
| I | 16 (24,6) |
| II | 41 (55,4) |
| III | 17 (23) |
| Cobertura de salud, n (%) | |
| Sí | 54 (73) |
| No | 12 (16,2) |
| Incluir salud | 8 (10,8) |
| Procedencia, n (%) | |
| GBA | 40 (54,1) |
| Interior | 28 (37,8) |
| CABA | 5 (6,8) |
| Otros países | 1 (1,4) |
| Escolaridad, n (%) | |
| Común | 51 (68,9) |
| No escolarizado | 18 (24,3) |
| Especial | 4 (5,4) |
| Domiciliaria | 1 (1,4) |
| Internación domiciliaria, n (%) | 9 (12,1) |
AME: atrofia muscular espinal; CABA: Ciudad Autónoma de Buenos Aires; DE: desviación estándar; GBA: Gran Buenos Aires; IQR: rango intercuartílico.
Con relación a la subclasificación, de los pacientes AME 1 (n=16) 13 (81,2%) fueron 1-B y tres (18,7%) 1-C. En el grupo AME 2 (n=41) 34 (82,9%) fueron A y siete (17%) B. Por último, del grupo AME 3 (n=17) 15 (88,2%) fueron A y dos (11,7%) B.
Se reportó el número de copias del SMN2 en 22 sujetos (29,7%), cinco de ellos (22,7%) tuvieron dos copias, 14 (63,6%) presentaron tres copias y tres (13,6%) cuatro copias. En la tabla 2 se presentan las variables clínicas de la muestra distribuidas por tipo de AME.
Características de los pacientes según su tipo de AME
| Variables | AME 1 | AME 2 | AME 3 |
|---|---|---|---|
| n=16 | n=41 | n=17 | |
| Edad de inicio de los síntomas, mediana (IQR), meses | 4 (2-5) | 8 (5-11)a | 23 (18 - 24)e |
| Edad de diagnóstico, mediana (IQR) meses | 6 (4,2-7,7) | 16 (12-24)b | 36 (24,5 - 48)f |
| Tiempo hasta el diagnóstico, mediana (IQR), meses | 2 (1-3,7) | 7 (2-14)c | 15 (6 - 28)e |
| AKR, n (%) | 16 (100) | 38 (92,6) | 11 (64,7) |
| Soporte ventilatorio, n (%) | 13 (81,2) | 17 (42,5)d | 1 (5,8) |
| Acceso a equipamiento, n (%) | 14 (87,5) | 37 (90,2) | 16 (94,1) |
| AKM, n (%) | 16 (100) | 34 (82,9) | 5 (29,4) |
| Escoliosis, n (%) | 16 (100) | 34 (82,9) | 8 (47) |
| Contracturas en MMII, n (%) | 4 (25) | 27 (65,8) | 2 (11,7) |
| Articulaciones afectadas, mediana (IQR), núm. | 1,5 (1-3,5) | 2,50 (2-4) | 3 (2 - 4) |
| Internaciones por intercurrencias respiratorias, n (%)* | 5 (31,2) | 9 (21,9) | 0 (0) |
| Nusinersen, n (%) | 11 (68,7) | 25 (60,9) | 4 (23,5) |
AKM: asistencia kinésica motora; AKR: asistencia kinésica respiratoria; AME: atrofia muscular espinal; DE: desviación estándar; IQR: rango intercuartílico; MMII: miembros inferiores; núm.: número.
Se estudió el estado motor actual con relación al tipo de AME, observándose que 23 (31,1%) sujetos perdieron logros motores que habían alcanzado, cuatro (5,4%) mejoraron sus habilidades motoras, mientras que 47 (63,5%) mantuvieron el mismo nivel funcional esperable para su fenotipo. En la figura 2 se pueden observar los máximos logros motores y los reportados en 2021, siete sujetos empeoraron, 29 se mantuvieron estables y todos los pacientes que habían mejorado su condición motora habían recibido nusinersen como tratamiento farmacológico.

Sobre los pacientes que lograron la marcha con o sin asistencia (n=28), 15 (53,57%) perdieron dicha capacidad reportándose una media de 63,1 meses (DE 44,1). Entre los pacientes que preservaron la marcha hasta la última evaluación, dos pertenecieron al grupo AME 2 (18,2%) y 10 (81,8%) al grupo de AME 3.
De los participantes que tenían escoliosis, 11 (14,9%) fueron sometidos a cirugía, todos del grupo AME 2. Otros resultados con relación al perfil motor se presentan en la tabla 3.
Perfil motor, respiratorio, tratamiento farmacológico, independencia y alimentación
| Otros resultados | |
|---|---|
| Variables | Todos(n=74) |
| Perfil motor | |
| Acceso a equipamiento, n (%) | 67 (90,5) |
| Acceso a AKM, n (%) | 55 (74,3) |
| ≤ 3 veces por semana | 38 (69) |
| >3 veces por semana | 17 (30,9) |
| Perfil respiratorio | |
| Soporte ventilatorio, n (%)* | 31 (42,4) |
| Causas de soporte ventilatorio, n (%) | |
| Intercurrencias respiratorias a repetición | 9 (12,1) |
| Falla de extubación o ARM prolongado | 9 (12,1) |
| Hipoventilación alveolar | 5 (6,7) |
| SAOS | 3 (4) |
| Prequirúrgico de API | 1 (1,3) |
| Terapia de higiene bronquial, n (%)** | |
| Técnica de airstacking | 32 (48,4) |
| Asistente mecánico de la tos | 32 (48,4) |
| AKR convencional | 1 (1,5) |
| AMT | 1 (1,5) |
| Pacientes con cánula de TQT, n (%) | 11 (14,8) |
| Tratamiento farmacológico | |
| Nusinersen, n (%) | 41 (55,4) |
| Dosis de nusinersen, mediana (IQR) núm. | 7 (4-10) |
| Salbutamol vía oral, n (%) | 25 (33,7) |
| Terapia génica, n (%) | 2 (2,7) |
| Actividades de la vida diaria | |
| Dependencia parcial, n (%)* | 45 (61,6) |
| Dependencia total, n (%)* | 22 (30,1) |
| Independientes, n (%)* | 6 (8,2) |
| Alimentación | |
| Vía oral exclusiva, n (%) | 57 (77) |
| SNG exclusiva, n (%) | 7 (9,4) |
| Gastrostomía, n (%) | 6 (8,1) |
| SNG para recuperación nutricional, n (%) | 4 (5,4) |
AKM: asistencia kinésica motora; AKR: asistencia kinésica respiratoria; AMT: asistencia manual de la tos; API: artrodesis posterior instrumentada; ARM: asistencia respiratoria mecánica; núm.: número; SNG: sonda nasogástrica; SAOS: síndrome de apnea obstructiva del sueño; TQT: traqueostomía.
Los resultados relacionados al perfil respiratorio se muestran en la tabla 3. De n=74, 66 pacientes (89,18%) realizaban algún tipo de terapia de higiene bronquial y 31 (41,89%) requerían asistencia ventilatoria no invasiva (VNI), 23 (31,08%) de forma parcial y ocho (10,81%) por más de 16 horas.
Tratamiento farmacológicoLos pacientes recibieron salbutamol oral, nusinersen y la terapia génica. Los resultados se reportan en la tabla 3. El más utilizado fue nusinersen, en un 55,4% de los casos.
DiscusiónEste estudio describe las características clínicas de una serie de pacientes con AME en seguimiento regular en un hospital pediátrico. Existen trabajos similares realizados en países limítrofes, no obstante, presentan muestras pequeñas, además de no poseer las mismas políticas de salud, lo que dificulta la comparación entre poblaciones8. A pesar de la baja prevalencia de esta patología, logramos reclutar 74 pacientes, ya que este estudio fue realizado en un hospital que es centro de referencia de seguimiento de pacientes neuromusculares.
En nuestra cohorte de pacientes al igual que en otros trabajos latinoamericanos predominaron los individuos pertenecientes al grupo AME 23,6. Creemos que esto podría relacionarse con la menor sobrevida del primer grupo5. Actualmente, con el diagnóstico más temprano y el acceso a las nuevas drogas modificadoras de la enfermedad, sería posible encontrarnos en futuros estudios con pacientes AME tipo 1 con una mayor sobrevida.
Con relación al diagnóstico, la bibliografía reporta la importancia de que este se realice de manera temprana, impactando en las posibilidades funcionales y la expectativa de los pacientes y sus cuidadores. Cao et al., reportan que los factores que podrían influir en su demora serían la diferencia entre la edad de sospecha de la enfermedad y su diagnóstico efectivo, es decir, el tiempo desde que la familia nota los síntomas y concurren a la consulta, el acceso a profesionales de salud especializados y a los estudios genéticos específicos12. Las publicaciones reportan una demora variable y amplia, de entre uno y 33 meses, sin discriminar según subtipo de AME3. En nuestra serie, el tiempo entre la sospecha y el diagnóstico fue menor. Se debe considerar que el inicio de los síntomas fue reportado por los padres y obtenido de las historias clínicas, por lo que podría ser un dato no certero. Además, ante la sospecha clínica, una vez que el pediatra deriva al paciente a un centro especializado, el diagnóstico suele facilitarse. Esto destaca el rol del pediatra de cabecera en la detección precoz de los signos y síntomas de la enfermedad y la derivación oportuna.
La bibliografía describe cómo se comporta esta patología en relación con las pérdidas de pautas motoras que se dan inevitablemente por evolución natural de la enfermedad13. Las guías internacionales recomiendan la implementación de kinesioterapia motora y la administración de drogas modificadoras de la enfermedad, con el objetivo de frenar el avance de la misma1,14,15. En nuestro estudio, más de la mitad mantuvo el máximo logro motor para su subtipo de AME. Dentro del grupo AME 1, dos sujetos lograron la sedestación independiente luego de recibir tratamiento con nusinersen, esta mejoría motora también fue reportada en otros estudios16–18, y aunque sería interesante describirlo en nuestra población por las características muestrales como por el tipo de trabajo, esto excede a los objetivos del presente. Con respecto a los pacientes del grupo AME 3, nueve perdieron la marcha independiente alrededor de los cinco años. En un estudio publicado en 2021 por Lusakowska que incluyó 393 pacientes con diagnóstico de AME tipo 3, la edad reportada de pérdida de marcha fue de 10 años aproximadamente, diferencia que podría deberse a que nuestra serie solo incluyó 17 pacientes con este subtipo15.
El abordaje kinésico se basa en prevenir y tratar las complicaciones ortopédicas y respiratorias. La escoliosis es una de las más frecuentes, esto coincide con los datos encontrados en nuestra serie donde más de la mitad de los pacientes la presentaron1,4. Los que requirieron cirugía pertenecieron en su totalidad al grupo 2. Posiblemente, el manejo quirúrgico en este subgrupo obedece a la necesidad de sostener un sentado adecuado en la silla de ruedas para favorecer su integración social y escolar.
Dentro de los cuidados recomendados, el otro pilar lo constituye el abordaje respiratorio, basado en el entrenamiento de las familias y los pacientes en terapias de higiene bronquial y/o la implementación de VNI precoz y oportuna14,19. Encontramos que la mayoría de los individuos contaban con entrenamiento familiar en alguna terapia de higiene bronquial, y los eventos respiratorios fueron poco frecuentes. Por ello podríamos inferir que estas intervenciones respiratorias serían un factor al menos mitigante en relación con las intercurrencias, información que coincide con lo indicado por guías internacionales14. Tanto en la bibliografía como en nuestra serie, la VNI fue el método preferente para soporte ventilatorio20,21. A pesar de ello, 11 niños estaban traqueostomizados, nueve con AME 1 y ventilación permanente. Si bien hay reportes de la utilización de VNI en pacientes con AME 1, en nuestro medio sigue siendo dificultoso el acceso precoz y oportuno para estos pacientes con severo compromiso bulbar.
En cuanto a la accesibilidad al sistema de salud, la mayor parte contaba con cobertura ya sea a través de obra social o prepaga. La mayoría poseía el equipamiento correspondiente y realizaba tratamiento kinésico de manera regular. A pesar del costo de las nuevas drogas modificadoras de la enfermedad, más de la mitad pudo acceder a nusinersen, reportándose una mediana de siete dosis por paciente. En estudios realizados en Latinoamérica no encontramos reportes de estas variables, posiblemente debido a las diferencias en los sistemas de salud de cada país y a las dificultades para acceder a medicamentos de alto costo.
Las limitaciones de este estudio se debieron al carácter retrospectivo del mismo, con pérdida de datos e imposibilidad de contar con información adicional o diferente a la registrada en las historias clínicas digitales. El menor número de pacientes dentro de los tipos 1 y 3 no permite generalizar los hallazgos en estos grupos. Asimismo, consideramos que la cantidad de pacientes pudo verse afectada ya que en el contexto de la pandemia de COVID-19 muchos descontinuaron sus controles y perdieron citas programadas.
Desde nuestro conocimiento, este es el primer estudio realizado en nuestro país en el que se describen las características de los pacientes con AME en seguimiento regular en un centro de referencia. Las variables estudiadas y los resultados de este trabajo revelan la complejidad en el abordaje de los pacientes con esta patología y la necesidad del trabajo multidisciplinario, en el cual los kinesiólogos cumplimos un papel relevante. El objetivo a futuro es generar redes de detección y tratamiento a lo largo de nuestro país. El desafío es replicar estos equipos de trabajo interdisciplinario en diferentes centros de atención que sean extensivos a todo el territorio nacional, que permitan un manejo proactivo de la patología con la consecuente descentralización de la atención.
Consideramos al presente trabajo como el punto de partida para nuevos estudios que abarquen intervalos de seguimiento más amplios, así como la incorporación de otros centros para lograr trabajos prospectivos multicéntricos.
ConclusiónNuestra muestra incluyó 74 pacientes con diagnóstico molecular confirmado de AME, la mayoría pertenecientes al grupo AME 2 de género masculino. El tiempo hasta el diagnóstico fue menor en comparación con estudios reportados en Latinoamérica. En su mayoría se trataba de pacientes provenientes del Gran Buenos Aires que contaban con cobertura en salud y que se encontraban escolarizados. En cuanto al perfil funcional, la mayoría mantuvo el logro motor máximo esperable para su fenotipo.
En lo que respecta a lo respiratorio, todos los pacientes que lo requirieron contaban con entrenamiento en alguna técnica de asistencia para la tos. La VNI fue el método predominante de ventilación dentro de nuestra muestra. La incidencia de internaciones debidas a causas respiratorias fue baja.
En relación con el tratamiento, tanto de rehabilitación como farmacológico, se encontró que la mayoría contó con acceso a los mismos, aún a pesar del alto costo de este último. En cuanto al equipamiento casi la totalidad de los pacientes contaron con lo que necesitaban según su estado motor.
Finalmente, consideramos que es fundamental destinar recursos para realizar nuevos estudios de carácter multicéntrico y prospectivo para caracterizar esta población en el particular contexto socioeconómico de nuestro país, así como en la formación del personal de salud en el abordaje multidisciplinario e integral de la enfermedad para lograr la pesquisa de los signos clínicos de manera temprana.
Declaración de la inteligencia artificial generativa y las tecnologías asistidas por inteligencia artificial en el proceso de escrituraLas autoras declaran no utilizar herramientas de inteligencia artificial (IA) durante ninguna fase del proceso de realización del presente trabajo.
FinanciaciónEste trabajo no ha recibido ningún tipo de financiación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
