




Hairy cell leukemia (HCL) is an uncommon B-cell lymphoid neoplasia, representing 2–3% of all leukemias. It is more common in men, with a median age at diagnosis of 52 years. The hair-like projections on its surface are its principal characteristic. Although its etiology has not been established, the recent gene sequencing of HCL identified the presence of the BRAF V600E mutation, absent in other malignant neoplasias of the lymph cells. The clinical course of the disease is usually indolent. The majority of patients initially present weakness and fatigue, pancytopenia and splenomegaly.
HCL must be distinguished from other indolent lymphoid neoplasias such as prolymphocytic leukemia, splenic marginal zone lymphoma, the variant of HCL (HCLv), and mantle cell lymphoma. Peripheral blood flow cytometer is essential for the detection of CD11c, CD19, CD20, CD22, CD25, and CD10, as well as a bone marrow aspirate to detect immunophenotypes. In addition, a bone biopsy is useful to perform an immunohistochemistry analysis for TRAP, DBA-44 and A1 annexin.
Purine analogues remain the first line of treatment. However, interferon and rituximab are a valid option, if the ideal treatment is unavailable. New discoveries in the pathophysiology of HCL have brought the creation of pharmaceuticals with distinct therapeutic targets. These pharmaceuticals are currently undergoing testing.
Hairy cell leukemia (HCL) is a B-cell lymphoid neoplasia. HCL differentiates from other B-cell neoplasms because of its morphological, immunophenotypic and molecular characteristics. Its main characteristic is the accumulation of monoclonal B cells, with hair-like projections on its surface, mainly found in peripheral blood, bone marrow and the spleen. It was described in 1958 by Bournoncle et al. However, they called it leukemic reticuloendotheliosis.1 The term “hairy cells” was coined in 1996 by Schrek et al.2 Today it is classified by the World Health Organization as a B-cell non-Hodgkin lymphoma.3
DefinitionHCL is a rare, chronic, lymphoproliferative B-cells disorder. Its main characteristics are the cytoplasmic prolongations which give the cells a hairy aspect.
EtiologyAlthough there are different studies showing a link between exposure to certain agents and HCL, its etiology has not been clearly established, which could be explained due to its low incidence. Amongst the agents which have proven a positive link are: exposition to pesticides,4 herbicides, mineral oils, working as a carpenter, or a farmer.4–7 Recently, a positive link with size has been described,5 whereas smoking has an inverse association, especially in men.8 The specific mechanism which confers the protection is unknown; nevertheless, smoking has been suggested to decrease the severity of inflammatory mechanisms9 and that nicotine induces apoptosis in lymphocytes.10
EpidemiologyHCL represents 2–3% of all leukemias. There are 600 new cases reported per year in the U.S. (3.2 cases per million inhabitants). Median age at diagnosis is 52 years old and it is more common in men than in women, with a 4:1 ratio; with a higher incidence in the white population, especially among Ashkenazi Jews.3 In Mexico, HCL represents 1.12% of all leukemias. However, in the north of the country it represents up to 1.83%, similar to the information from the U.S.11,12
PhysiopathologyHCL is a chronic, lymphoproliferative B-cell disorder. However, its cells do not have the appearance of any B-cell sub-population, and its origin has been a matter of debate. The analysis of immunoglobulins (Ig) variable regions genes is a tool used to discover the lymphoid cells’ clonal origin.13 In over 85% of cases, we are able to find somatic mutations in Ig variable regions genes of HCL cells,14,15 which is an indication that the cells involved have passed through the germinal center or lymphoid peripheral organs.16 Approximately 40% of HCL cells co-express multiple clonally-related Ig isotypes.17
Evidence suggests an origin post germinal center in memory B-cells, due to their genomic expression profile.18,19 The origin of memory B-cells is compatible with HCL's lack of chromosomic reciprocal translocation.20 Absence of CD27 is typical in HCL. This represents a point against the hypothesis of its origin in memory B-cells.21 However, negative CD27 memory B-cells have been observed.22 Since lymph node affection is infrequent, it has been suggested that the HCL origin cell is probably located in the bone marrow or spleen, since those are usually the more affected places. HCL cells show an expression profile similar to the spleen's marginal zone.23
HCL's characteristic look is due to the beta-actin expression, which is polymerized to F-actin, located in the cortical cytoskeleton.24 PP52 phosphoprotein, which is specific for leukocytes, is linked to F-actin and is responsible for bringing support to the hair-like projections.25 On the other hand, HCL gene sequencing recently identified the presence of a BRAF V600E mutation in almost every patient with the disease, absent in other B-cell lymphoid malignancies.26,27 BRAF mutations activate the MAPK pathway, promoting growth, survival and HCL cell differentiation.28
Clinical presentation and lab findingsThe disease's clinical course is indolent. Most patients usually present weakness and fatigue as the predominant symptoms during the onset of the disease.29 Sometimes, there is a history of repeated infections. The fisical examination findings are: splenomegaly in 96%, hepatomegaly in 58% and lymphadenopathy in only 35%. These swollen lymph nodes are rarely observed in the periphery; nonetheless, they are generally present in the abdomen and detected by imaging studies.30
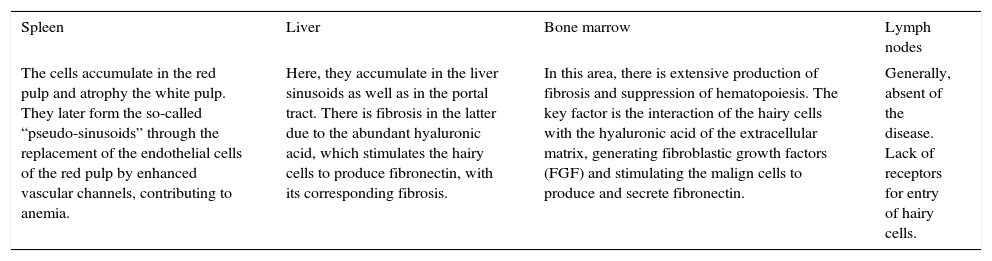
In an advanced stage of the disease we are able to find pain in the superior left quadrant, infections, fever, and hemorrhages and/or weight loss. However, this is uncommon because of the availability and effectiveness of treatment.30,31 Clinical manifestations are product of hairy cell accumulation in the spleen, liver and bone marrow (Table 1).32
Clinical manifestations.
| Spleen | Liver | Bone marrow | Lymph nodes |
|---|---|---|---|
| The cells accumulate in the red pulp and atrophy the white pulp. They later form the so-called “pseudo-sinusoids” through the replacement of the endothelial cells of the red pulp by enhanced vascular channels, contributing to anemia. | Here, they accumulate in the liver sinusoids as well as in the portal tract. There is fibrosis in the latter due to the abundant hyaluronic acid, which stimulates the hairy cells to produce fibronectin, with its corresponding fibrosis. | In this area, there is extensive production of fibrosis and suppression of hematopoiesis. The key factor is the interaction of the hairy cells with the hyaluronic acid of the extracellular matrix, generating fibroblastic growth factors (FGF) and stimulating the malign cells to produce and secrete fibronectin. | Generally, absent of the disease. Lack of receptors for entry of hairy cells. |
Respect to lab studies, it is frequent to observe anemia in 85%, thrombocytopenia in 60.80% and leukopenia in 60% due to hypersplenism and bone marrow infiltration.30
Differential diagnosisHCL must be differentiated from other indolent lymphoid malignancies such as prolymphocytic leukemia, splenic marginal zone lymphoma, mantle cell lymphoma and HCL variant (HCL-v). The last one occurs in 10% of cases with a median age of 70 years, and despite the similarities with classic hairy cell leukemia, they diverge in the absence of the CD25 and CD123 immunophenotypic markers. Another way of making a differential diagnosis is the lack of response to standard HCL treatment and the nonexistence of mutations of the BRAF V600F gene.30
Diagnostic methodsHCL diagnosis is commonly made with a biopsy and bone marrow aspiration combined with immunophenotypic characterization through flow cytometry.33 It is important to point out that this pathology is usually sub-diagnosed and requires clinical suspicion and the use of the proper technology to solve this problem. As previously mentioned, most patients (70–90%) present pancytopenia, with leucopenia (<5×109/L), anemia (<10g/dL), neutropenia (<1×109/L), monocytopenia (<0.1×109/L) and thrombocytopenia (<100×109/L). Only between 10% and 20% present moderate leukocytosis (>10×109/L). HCL patients display elevated IL-2R (CD25) serum levels, which correlates with the disease's degree of activity.34 Other tests which should be considered when making the diagnosis are serum immunoglobulin levels, as well as IgVH gene and BRAF V600E somatic mutations.32 Some HCL histopathologic and immunophenotypic characteristics are the following:
- •
Lymphocyte flow cytometry in peripheral blood or bone marrow with CD19, CD20, FMC7, CD11c, CD103, CD25, HC2, CD22, sIg, CD79a and CD123 expressions, with four being the main and specific markers: CD11c, CD103, CD25 and CD123.34 Commonly negative markers are CD5, CD23, CD10, CD79b and CD27.32
- •
A strong expression for CD200 is characteristic of HCL and may be useful in the diagnosis of difficult cases.34
- •
Bone marrow aspiration with a needle may be difficult to obtain and is frequently unproductive or “dry”. In a bone marrow biopsy, we can observe fibrosis, with a cellular “fried-egg” look caused by the wide spaces between nuclei and abundant cytoplasm. Immunohistochemistry analyses for CD20 and TRAP (tartrate resistant acid phosphatase), DBA-4 and annexin A1 are conducted, which are characteristically positive.32
Andrulis et al. directed a study where the efficiency of the VE1 antibody for BRAF V600E detection was reported, along with HCL identification in other entities. Moreover, a study conducted by Uppal et al. found a sensitivity of 88% and a specificity of 97% to detect this mutation with the mentioned antibody.34
Current treatmentHCL regularly has an indolent evolution. Waiting and observing is a good option for asymptomatic patients, since early treatment does not offer any sort of benefit in the survival rates of these cases. Anyway, the progression of the disease in most patients will lead to complications as a result of cytopenias and splenomegaly; i.e. anemia, hemorrhages, recurrent infections, etc. In general practice, treatment must be started if any of the criteria listed in Table 2 occur.35 When the decision to not start treatment is taken, clinical and laboratory monitoring should be done for every three months during the first year and every six months thereafter.35 HCL treatment is not considered healing; but current treatment strategies are capable of reaching prolonged remissions, thus increasing global survival rates (Algorithm 1).36–39

In 1960, findings showed that 30% of children with severe combined immunodeficiency syndrome lacked deaminase adenosine enzyme (ADA).40 Furthermore, they discovered that the accumulation of the deoxyadenosine triphosphate form was responsible for the decrease of lymphocytes.41 Keeping these observations in mind, medications capable of binding irreversibly to ADA or antagonizing its action were developed. After treatment with purine analogues, deoxyadenosine triphosphate accumulation results in rupture and inhibition of the DNA repair, which translates into cellular apoptosis.42
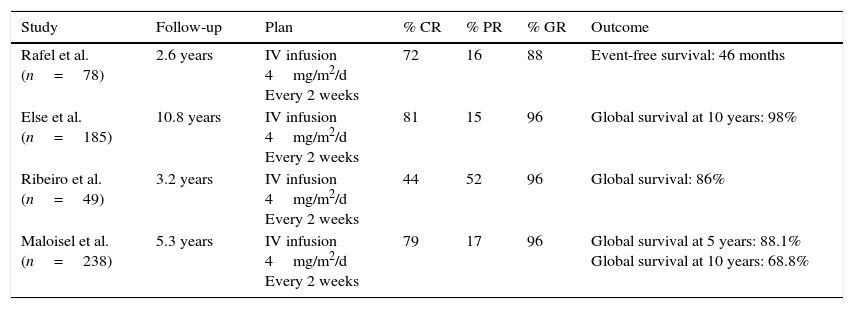
PentostatinAlso known as 2′-deoxycoformycin (dcF), a product of Streptomyces antibioticus and ADA inhibitors, first introduced in 1980 as the first purine analogues for HCL treatment. In Table 3,37,39,43,44 specific studies show response rates to pentostatin. Intravenous use of pentostatin at a 4mg/m2 ratio once every two weeks until reaching complete response was approved in the U.S. Pentostatin is safe in patients with creatinine lightening >60mL/min. However, a dosage reduction is necessary if this depuration is between 40 and 60mL/min.32 Patient hydration with 1.5L of intravenous solution with every pentostatin cycle is recommended.30
Studies on the effectiveness of pentostatin in HCL cases.
| Study | Follow-up | Plan | % CR | % PR | % GR | Outcome |
|---|---|---|---|---|---|---|
| Rafel et al. (n=78) | 2.6 years | IV infusion 4mg/m2/d Every 2 weeks | 72 | 16 | 88 | Event-free survival: 46 months |
| Else et al. (n=185) | 10.8 years | IV infusion 4mg/m2/d Every 2 weeks | 81 | 15 | 96 | Global survival at 10 years: 98% |
| Ribeiro et al. (n=49) | 3.2 years | IV infusion 4mg/m2/d Every 2 weeks | 44 | 52 | 96 | Global survival: 86% |
| Maloisel et al. (n=238) | 5.3 years | IV infusion 4mg/m2/d Every 2 weeks | 79 | 17 | 96 | Global survival at 5 years: 88.1% Global survival at 10 years: 68.8% |
RC: complete response, RP: partial response, RG: global response.
Global response varies between 88% and 96%, while complete response is between 44% and 81%. Flinn et al. estimated a survival rate of 5 and 10 years of 90% and 81%, respectively, with a mean follow-up duration of 9.3 years.45 Pentostatin is generally well-tolerated and the most common adverse effects are anemia, thrombocytopenia and neutropenia.46 Still, pentostatin has been reported to decline CD4+ and CD8+ lymphocyte counts significantly, which could increase secondary malignancies and incidence of infections.47,48
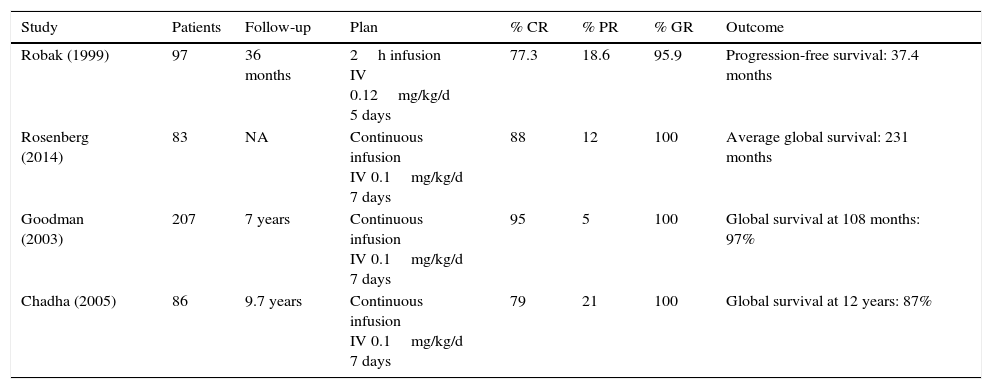
CladribineIt is known as 2-chlorodeoxyadenosine (CdA). In Table 4,49–52 studies indicate its effectiveness. The most used scheme consists of administering 0.1mg/kg/day in a continuous infusion for 7 days. In a non-randomized study, there was evidence proving that there was no statistically significant difference in the response and toxicity ranges between infusions (24h and 2h).53 Another randomized study compared daily versus weekly cladribine administration; there were no significant findings in response, survival, global and toxicity rates.54 A different study showed that the weekly program reduced the risk of infections.55 One of the advantages of subcutaneous administration is the fact that in most cases it does not require hospitalization. 0.14mg/day for 5 days has a 95% response rate,56 similar to the intravenous administration. Weekly subcutaneous programs have similar response and toxicity rates as daily ones.57 With just one cladribine cycle, a global response up to 100% can be obtained, and total response rates differ from 77% to 95%.49–52 Jehn et al. reported a global survival at 12 years of 79%.36 In general, cladribine is well-tolerated, with cytopenias and fever being the most common adverse effects.
Studies on the effectiveness of cladribine in HCL cases.
| Study | Patients | Follow-up | Plan | % CR | % PR | % GR | Outcome |
|---|---|---|---|---|---|---|---|
| Robak (1999) | 97 | 36 months | 2h infusion IV 0.12mg/kg/d 5 days | 77.3 | 18.6 | 95.9 | Progression-free survival: 37.4 months |
| Rosenberg (2014) | 83 | NA | Continuous infusion IV 0.1mg/kg/d 7 days | 88 | 12 | 100 | Average global survival: 231 months |
| Goodman (2003) | 207 | 7 years | Continuous infusion IV 0.1mg/kg/d 7 days | 95 | 5 | 100 | Global survival at 108 months: 97% |
| Chadha (2005) | 86 | 9.7 years | Continuous infusion IV 0.1mg/kg/d 7 days | 79 | 21 | 100 | Global survival at 12 years: 87% |
RC: complete response, RP: partial response, RG: global response.
Until now, there are no randomized prospective studies comparing pentostatin versus cladribine, in part because of the great efficiency of both drugs and due to low HCL incidence. Even so, there are retrospective studies proving the fact that both drugs have a similar efficacy, in terms of complete response and free-of-disease survival.
Other treatmentsPurine analogues remain the first line of treatment but new discoveries regarding HCL pathophysiology have led to the creation of drugs with different therapeutic targets. These drugs are under research and some have shown promising results.
RituximabSince HCL is a B-cell malignancy, it is logical to use a monoclonal antibody against CD20, such as rituximab. Employed as a stand-alone drug, rituximab may reach total response rates of 10–54% in patients with an HCL relapse, at a 375mg/m2 dosage once a week for 4–8 weeks.58,59 Else et al. retrospectively reviewed 18 patients who were treated with purine analogues in combination with rituximab as a second line of treatment, after being treated with purine analogues as single agents. All patients responded, with a complete response rate of 89%.60
Rituximab at 375mg/m2 per week for 8 weeks as initial therapy after administrating 5.6mg/m2 cladribine via 2-h IV infusion for 5 days generates a total response of 100%.61 In special or unfavorable situations, 100mg of rituximab per week may be utilized for 4–6 weeks. This is less expensive and is usually effective, especially when is combined with interferon.
While purine analogues may not be capable of eliminating HCL, since minimal residual disease (MRD) detected after cladribine administration is always strongly CD20+, MRD eradication may be obtained using rituximab. Rivandi et al. proved in a preliminary study that rituximab at conventional doses for a period of 8 weeks performs with great activity, eliminating MRD in 13 patients, when it is used 4 weeks after the administration of cladribine.62
VemurafenibAs described earlier, BRAF V600E mutation is the genetic key in HCL. Therefore, it is a therapeutic target which has been studied in recent years. Vemurafenib is a BRAF V600E oral inhibitor. Tiacci et al. conducted a study to measure Vemurafenib activity and safety in patients with HLC who relapsed after treatment with purine analogues or who were refractory to the administration of purine analogues. Global response rate was 96% and complete response rate was 35%, with a relapse-free survival mean of 19 months. The adverse effects were rash, arthralgia and arthritis.63
Since a positive relation has been observed between the use of Vemurafenib and the onset of dermatological malignancies, frequent explorations of the skin are recommended.
IbrutinibA selective and irreversible inhibitor of the Burton's tyrosine kinase intervenes in the B-cell signaling pathway.64 An ibrutinib clinical assay in patients with HCL relapse has recently begun. Preliminary efficacy and safety data show adverse effects such as eruptions, diarrhea and arthralgia. This clinical assay is currently taking place in several centers in the U.S. (NCT01841723).
ImmunotoxinsIn order to increase monoclonal antibody cytotoxicity, techniques that facilitate the production of antibody–toxin or antibody–drug conjugates have been created. An immunotoxin is the fusion between a bacterial toxin (i.e. Pseudomonas exotoxin or diphtheria) and the variable fraction of a monoclonal antibody, whose specific target is found on the surface of neoplastic cells like CD25 or CD22. This toxin is released in the neoplastic cell's interior and interferes with protein synthesis.65
BL22 is an immunotoxin against CD22 fused with a truncated shape of the P. exotoxin PE38. In a phase II clinical assay, BL22 was tested in 36 HCL relapse or refractory disease cases. After a cycle (40mg/kg every two days, three doses), complete response rate was 25% and global response rate was 50%. These responses improved to a total response rate of 47% and a global response rate of 72% after retreatment (only in patients with cytopenias). Two patients developed uremic hemolytic syndrome without the need to recur to plasmapheresis.66 Subsequently, moxetumomab pasudotox was developed as a modified version of BL22 with a higher affinity and cytotoxicity. In a phase I assay, which included 28 patients with HCL relapse and resistance, a global response rate of 86% was obtained, including a sustained complete response rate in 46% of patients.67
Therapeutic options in unfavorable circumstancesEven though HCL is treated in most developed countries with cladribine and pentostatin, it is a fact that these drugs are not only expensive, they are not available in Mexico and in many countries with limited resources. So, in these types of circumstances, there are other affordable therapeutic options with favorable results
Interferon alpha for HCL patient treatment was first introduced in 1984. Today, its use is limited, mainly due to purine analogues’ great effectiveness. On the other hand, in countries with low economic resources, it is an inexpensive option and one which has proved similar results to those of cladribine in terms of global survival. Ruiz-Delgado et al. conducted a comparative study between interferon alpha (n=18) and cladribine (n=11), where the difference in global survival between both groups was not statistically significant; 94% at 217 months in the interferon group and 91% at 133 months in the cladribine group.68 In a study conducted in our center, nine HCL patients received three IFN mega-units three times a week for 12 weeks, subsequently they received treatment once again for 8 weeks when there was a leukemia reactivation or after 10 months of observation each year. All patients had a hematological remission before 12 weeks of treatment. This therapeutic option is cheaper, effective and comparable to other forms of therapy with IFN in the treatment and maintenance of patients with this type of leukemia.69 It is possible to combine interferon with rituximab without increasing toxic effects and improving effectiveness.
Splenectomy was the first intervention that significantly changed the survival in patients. Today, it is rarely used. It may be recommended in patients with painful massive splenomegaly (>10cm under the costal edge) and with minimal infiltration of the bone marrow, or in patients refractory to treatment with interferon and purine analogues.33 Retrospective studies show a complete response rate of 40–62%, and a mean survival rate at 5 years up to 68%.70,71 Lad et al. published a retrospective study, including 24 patients with an HCL diagnosis, they were divided into two groups: 17 patients received cladribine and 7 were splenectomized. 75% of patients in the splenectomy group showed total remission, 94% did so in the cladribine group. An interesting finding when comparing both groups was that no statistically significant differences were observed regarding leukemia-free survival and global survival.72
PrognosisSurvival time in patients after diagnosis was 4 years before a treatment was known, due to complications derived from cytopenias, including hemorrhages and infections. Thereafter, with splenectomy as a first line treatment, there was a complete response of 40–62% and a survival rate of 5 years at 61–68%. Then, alfa interferon was utilized as the first drug with benefits in the treatment of HCL. Still its full response rate was low, at 10%.73
Today, with purine analogues (pentostatin and cladribine), complete response is induced up to 80% of patients with a median survival of 10 years. Global response rates are 96–100% with a complete response rate of 80% and a survival rate of 10 years ranging between 85% and 100%.74 Despite that, a significant proportion of patients with HCL fail in their response to treatment or become resistant. Up to 48% of patients relapse in the following 15 years.75 The future of HCL patients is very favorable. The challenge is to identify this malignancy as early as possible and treat it properly using available resources.
Conflict of interestThe authors have no conflicts of interest to declare.