
















Los trastornos neurocutáneos (TNC) son afecciones generalmente heredofamiliares que se caracterizan por la presencia de alteraciones de estructuras de origen ectodérmico, afectando especialmente a la piel y al sistema nervioso1.
Históricamente han recibido diferentes denominaciones. El término facomatosis fue introducido a principios del siglo xx para englobar la esclerosis tuberosa y la neurofibromatosis tipo 1. Posteriormente estas designaciones han sido reemplazadas por neuroectodermomas, hamartosis o neuroectodemesodermosis. En la actualidad se emplea la expresión trastornos o síndromes neurocutáneos para englobar todos estos procesos.
La mayor parte de estos cuadros clínicos cumple unas características anatomopatológicas, genéticas y clínicas comunes. Las alteraciones observadas tienen una clara naturaleza displásica de inicio en fases precoces del desarrollo embrionario. Estas lesiones congénitas muestran un gran potencial de sobrecrecimiento con la aparición de hamartomas y tumores, benignos o malignos. Clínicamente pueden comprometer a numerosos órganos y la expresión sintomática es extremadamente heterogénea incluso dentro del mismo TNC y la misma familia. Una gran parte de estos trastornos tiene una base genética conocida, transmitida por uno de los progenitores o aparecida como una mutación de novo.
Los patrones hereditarios permiten clasificar estas enfermedades según se refleja en la tabla 12. Otras clasificaciones se basan en la lesión cutánea predominante (tabla 2)1.


Neurofibromatosis
El término neurofibromatosis designa un grupo de trastornos muy diversos que comparten algunas características pero que claramente difieren los unos de otros. Las dos formas más frecuentes son: la neurofibromatosis tipo 1 (NF1), enfermedad de Von Recklinghausen o subtipo periférico, y la neurofibromatosis tipo 2 (NF2) o neurofibromatosis central. Este último trastorno es infrecuente en la población infantil. Se hereda con carácter autosómico dominante y los criterios diagnósticos se exponen brevemente en la tabla 33.

Neurofibromatosis tipo 1 (NF1)4-6
Constituye el 85% de todos los casos de neurofibromatosis; es el TNC más frecuente. Su prevalencia se sitúa en 1/3.000 personas. Se hereda por vía autosómica dominante en el 98% de penetrancia aunque con gran variabilidad fenotípica. El 30-50% de los casos son nuevas mutaciones y por lo tanto, esporádicos.
El gen transmisor de la NF1 se sitúa en el cromosoma 17q11.1. Este gen codifica un segmento de la proteína neurofibramina, al que se le supone una acción supresora tumoral.
Alteraciones cutáneas
La manifestación cutánea más importante en la NF1 es las manchas café con leche (fig. 1). El 50% de los pacientes que muestran numerosas manchas café con leche presentará durante su desarrollo otras alteraciones diagnósticas de la NF1. Estas manchas tienden a aumentar en tamaño y número con la edad; aparecen predominantemente en axilas, ingles y cuello.
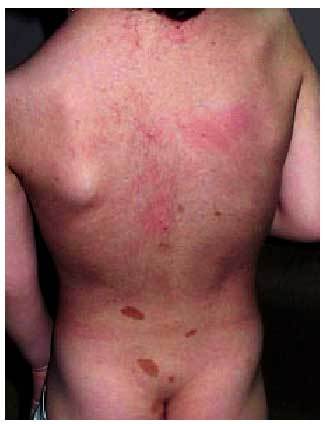
Fig. 1. Numerosas manchas café con leche en paciente con NF1.
Otra alteración característica es la presencia de neurofibromas dermosubdérmicos que se pueden observar en cualquier parte del cuerpo. Son generalmente asintomáticos; sin embargo, cuando se desarrollan en áreas de roce (manos, cintura, etc.) pueden ser dolorosos y precisar la extirpación quirúrgica. En el 30% de los casos se localizan en trayectos nerviosos, reciben el nombre de neuromas o neurofibromas plexiformes; cuando asientan en la región palpebral (5%), pueden alterar la agudeza visual.
Los nódulos de Lisch son hamartomas pigmentados en iris, específicos de la NF1. Se observan en el 25% de los casos antes de la pubertad, y en la mayoría de los pacientes a partir de la adolescencia.
Alteraciones neurológicas
La macrocefalia es frecuente (16-45%). Aunque puede asociarse con la presencia de hidrocefalia por estenosis del acueducto de Silvio, su origen se sitúa habitualmente en una megalencefalia asintomática.
La presencia de retraso mental es poco habitual (8%). El cociente intelectual medio en niños con NF1 se sitúa en 85, 15-20 puntos menos que sus hermanos no afectados. La presencia de dificultades de aprendizaje es elevada y fue descrita hasta en el 65% de los casos. Hasta en el 50% de los pacientes se objetiva hipercinesia y déficit atencional, que pueden responder al tratamiento estimulante.
La epilepsia afecta al 3,5-7,5% de los niños con NF1. Se han descrito todo tipo de crisis. El control terapéutico suele ser satisfactorio.
Entre las alteraciones neurológicas, la existencia de tumores intracraneales suele ser la mayor preocupación de los padres y profesionales. Los gliomas de vías ópticas son los más frecuentes, están presentes en el 15-20% de los niños afectados (fig. 2). En contrapartida,
el 50% de los gliomas ópticos se asocia con la NF1. Histológicamente se trata de astrocitomas pilocíticos benignos. Las manifestaciones clínicas varían desde la ausencia de síntomas (75%) hasta la presencia de proptosis y alteración de la agudeza visual. En la tercera parte de los casos puede asociarse con pubertad precoz. La aparición de síntomas ocurre generalmente antes de los 6 años, y rara vez a partir de la pubertad. El tratamiento de los gliomas de vías ópticas es controvertido; su escasa malignidad conduce a algunos especialistas a mantener una actitud expectante; la exéresis quirúrgica ha dado buenos resultados en algunos casos sintomáticos. En otros pacientes se ha empleado radioterapia y quimioterapia con una elevada frecuencia de efectos colaterales.
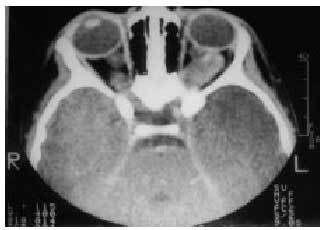
Fig. 2. TC craneal: glioma de nervio óptico en paciente con NF1.
Los tumores intracraneales restantes son infrecuentes y tienden a situarse en hemisferios cerebrales, cerebelo y tronco. Presentan una naturaleza histológica similar a la de los gliomas de vías ópticas. Cuando asientan en cerebelo o hemisferios cerebrales pueden ser tratados quirúrgicamente dependiendo de su volumen, crecimiento o repercusión clínica. Cuando se sitúan en el tronco cerebral recomendamos mantener una actitud mínimamente agresiva, por su escaso crecimiento y pobre asociación sintomática.
Otras alteraciones neurológicas menos frecuentes son la hidrocefalia secundaria a la estenosis acueductal (6%), malformaciones del sistema nervioso central (SNC) (agenesia del cuerpo calloso, hemimegalencefalia, polimicrogiria), tumores intraespinales y otras.

Alteraciones óseas
En el 3% de los pacientes se observa displasia facial, afectando predominantemente a la calota craneal, huesos maxilares y alas esfenoidales. Pueden manifestarse por asimetría facial o exoftalmus pulsátil.
La escoliosis es muy frecuente (20%), puede ser lo suficientemente grave como para afectar a la médula y necesitar corrección quirúrgica.
La seudoartrosis de huesos largos, especialmente la tibia, es común. Puede ser asintomática o sufrir fracturas frecuentes. Las intervenciones múltiples son habituales y el pronóstico desde el punto de vista funcional no suele ser favorable.
Otras manifestaciones
Otras alteraciones clínicas de la NF1 comprenden retraso del crecimiento (25%), hipertensión arterial (HTA) a veces secundaria a la estenosis de arterias renales, trastornos endocrinológicos especialmente la pubertad precoz, tumores viscerales nefroblastoma, leucemia y otros.
Diagnóstico
El diagnóstico de la NF1 se establece según los hallazgos clínicos, aunque el diagnóstico mediante genética molecular es posible.
Los criterios diagnósticos se recogen en la tabla 3.
Esclerosis tuberosa7-9
La esclerosis tuberosa (ET) o enfermedad de Bourneville es un TNC heredado de forma autosómica dominante con alta penetrancia y expresión variable, caracterizado básicamente por manchas acrómicas y tumores benignos o hamartomas. Tiene una prevalencia aproximada de 1-1,5/10.000 personas. El 60-70% de los casos son esporádicos. Se han identificado dos genes causantes, TSC1 y TSC2, localizados en los brazos cortos de los cromosomas 9 y 16 respectivamente, que actúan probablemente como supresores tumorales.
Alteraciones cutáneas
Su presencia es prácticamente diagnóstica de la enfermedad.
Las manchas acrómicas son lesiones hipomelánicas frecuentes (90%) y precoces (fig. 3). Son irregulares, de forma lanceolada, con bordes dentados y tamaños variables que recuerdan una hoja de fresno. Estas manchas son inespecíficas ya que se las puede observar hasta en el 1% de los recién nacidos sanos. Pueden adquirir otras formas o afectar al cuero cabelludo en forma de mechones blancos; es el primer signo de la enfermedad en algunos casos.
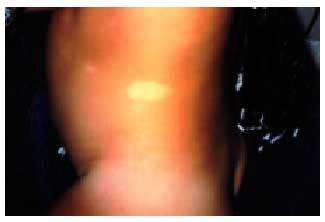
Fig. 3. Manchas acrómicas en paciente con ET.
Los angiofibromas faciales o adenomas sebáceos son lesiones características de la ET que asientan frecuentemente sobre la región nasogeniana (fig. 4). Se observan en el 50% de los niños y aparecen entre los 3 y los 15 años de edad.
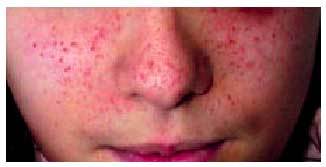
Fig. 4. Adenomas sebáceos en niña con ET.
Otros hallazgos dermatológicos habituales son la «piel de Marrasquino» (peau du chagrin) que se objetiva en la tercera parte de los pacientes, en forma de placas ligeramente elevadas, parduscas, predominantemente en el área lumbar. Los fibromas ungueales o tumores deKoenen son patognomónicos de la ET; se manifiestan en el 50% de los casos aunque rara vez se ven en la edad infantil. Las placas fibrosas son formaciones sobreelevadas que suelen observarse ya al nacimiento, asentadas sobre la frente o las mejillas.
Alteraciones neurológicas
La epilepsia es el trastorno neurológico más característico de la ET y afecta al 60-90% de los pacientes, generalmente en el primer año de vida. Las crisis pueden tener diferentes morfologías; las convulsiones más frecuentes son los espasmos infantiles, aunque también son frecuentes las crisis tónicas, atónicas o parciales complejas. Desde un punto de vista evolutivo, pueden adquirir las características de un síndrome de Lennox-Gastaut. La presencia de crisis convulsivas así como el control terapéutico precoz están estrechamente relacionados con el pronóstico cognoscitivo.
La manifestación de retraso mental se describe en el 50-80% de los pacientes. Esta situación se ha asociado con la presencia de túberes frontales, crisis parciales complejas y espasmos infantiles con mal control terapéutico.
Son igualmente frecuentes el autismo, los trastornos psicóticos, la conducta hipercinética o la agresividad, que se manifiestan en el 50% de los pacientes.
La ET, al igual que otros trastornos neurocutáneos, se asocia con tumores intracraneales. Los nódulos subependimarios son hamartomas característicos de este síndrome. Se observan generalmente calcificados, próximos al agujero de Monro, que pueden llegar a obstruirlo (6%). La presencia de tumores o astrocitomas gigantocelulares no supera el 5% de los casos; asientan en estructuras subependimarias, a menudo en la cabeza del núcleo caudado. La existencia de grandes calcificaciones en esta estructura y la captación de contraste en la tomografía computarizada (TC) cerebral son altamente sugestivas del desarrollo de estos tumores. La resonancia magnética (RM) cerebral revela con mayor precisión que la TC la presencia de tuberosidades corticosubcorticales, lesiones hamartomatosas íntimamente ligadas a la clínica.
Alteraciones oftalmológicas
Los hallazgos oculares están representados por los hamartomas o facomas de retina. Son astrocitomas benignos que se encuentran en el 50% de los pacientes, generalmente paucisintomáticos. Pueden ser únicos, múltiples, multinodulares en forma de mora y son fácilmente reconocibles cuando se calcifican. La presencia de cataratas, desprendimiento de retina o colobomas es excepcional.
Otras alteraciones
La afección multisistémica en la ET engloba asimismo alteraciones renales, cardíacas, pulmonares o digestivas.
En los riñones son frecuentes los quistes renales y angiomiolipomas, presentes en el 45-60% de los pacientes. Son habitualmente múltiples y bilaterales. Aunque suelen ser asintomáticos, pueden manifestarse por su hipercrecimiento, HTA, fallo renal o hemorragias.
Las manifestaciones cardíacas son también habituales. En el 50% de los niños se pueden encontrar hamartomas o rabdomiomas, comúnmente asintomáticos e involutivos. En algunos casos pueden obstruir las cavidades cardíacas, provocar una franca insuficiencia cardíaca, o arritmias cuando los rabdomiomas son intramurales.
La presencia de otras alteraciones, como aneurismas de la aorta descendente, linfangioleiomatosis pulmonar, pólipos rectales o alteraciones óseas, es menos habitual.
Diagnóstico
El diagnóstico se basa en los hallazgos clínicos descritos así como en otros radiológicos (cerebrales, cardíacos o renales). Estas manifestaciones se confirman como criterios diagnósticos (tabla 4).

Hipomelanosis de Ito o incontinentia pigmenti achromians10-13
Es el tercer TNC por orden de prevalencia; es superado exclusivamente por la NF1 y la ET. Se ha descrito una incidencia de 1-2/10.000 pacientes remitidos a consulta hospitalaria. Algunos autores apuntan incidencias probablemente superiores, motivadas por la falta de reconocimiento clínico del trastorno. Su base hereditaria es confusa. Se ha apuntado un patrón de herencia autosómico dominante, aunque se han descrito casos de origen probablemente recesivo o ligados al cromosoma X. En el 60% de los pacientes se puede llegar a confirmar alteraciones cromosómicas en cariotipos de sangre periférica o fibroblastos cutáneos; las más frecuentes son las trisomías 18 en mosaicismo, traslocaciones balanceadas de los cromosomas 2 y 8 u 8 y 16, mosaicismos de otros cromosomas (sexuales, 9, 14) o tetrasomía del 12.
Alteraciones cutáneas
Las lesiones hipopigmentadas son definitorias y deben hallarse en la totalidad de los pacientes. Se observan generalmente en la lactancia (70%), aunque rara vez al nacimiento. Son lesiones irregulares, de tamaño y localización muy variables, uni o bilaterales; en ocasiones adquieren figuras peculiares en forma de bandas, líneas en zigzag, formas geográficas, etc. La falta de pigmentación sigue con frecuencia las líneas de Blaschko (fig. 5).
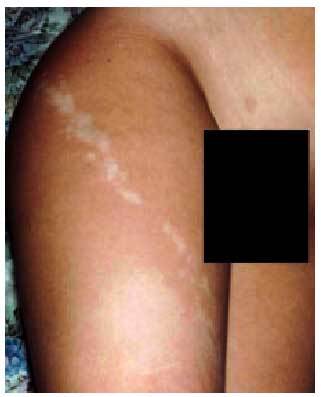
Fig. 5. Hipomelanosis segmentaria de Ito.
También son frecuentes otras alteraciones pigmentarias (38%) como las manchas café con leche, nevo de Ota, nevus marmoratus o manchas mongólicas persistentes.
Alteraciones neurológicas
Las alteraciones del SNC se describen en el 50-75% de los pacientes según diferentes casuísticas de control hospitalario. Según nuestra experiencia, la incidencia de estas alteraciones está sobredimensionada por las características metodológicas de las revisiones.
En relación con estas series, se observa retraso mental en el 70% de los casos, crisis convulsivas polimorfas en el 50% y autismo infantil en el 11% de los niños. Sin embargo, con frecuencia quedan infradiagnosticados pacientes asintomáticos, u otros con alteraciones neuropsiquiátricas de menor gravedad.
Aproximadamente el 20% de los niños muestra alteraciones neurorradiológicas que comprenden la atrofia corticosubcortical, las alteraciones de la sustancia blanca, los trastornos de la migración neuronal o la hipoplasia cerebelosa.
Alteraciones osteomusculares
La presencia de alteraciones musculares u óseas se objetiva en la tercera parte de los casos. Es frecuente la hipertrofia ipsilateral a las lesiones pigmentarias, las alteraciones esqueléticas subyacentes a las mismas, la asimetría de pectorales, los trastornos dentarios o las anomalías en la curvatura de la columna vertebral.
Otras alteraciones
Se han descrito numerosas alteraciones fenotípicas que expresan probablemente la variabilidad genotípica asociada. Así, se ha señalado la presencia de alteraciones oftalmológicas (heterocromía de iris, hipertelorismo, opacidad corneal, etc.), cardíacas (comunicaciones, Fallot), renales o genitales (micropene, criptorquidia).
Diagnóstico
El diagnóstico se basa exclusivamente en las características clínicas. Los hallazgos anatomopatológicos de las lesiones hipopigmentadas únicamente revelan la alteración inespecífica de los melanocitos epidérmicos, en relación con el número o el tamaño de los melanosomas. Los estudios genéticos son todavía poco concluyentes; los análisis cromosómicos deben ser realizados preferentemente en cultivos de queratinocitos, debido al elevado número de falsos negativos en los cultivos de linfocitos y fibroblastos.
Incontinentia pigmenti o síndrome de Bloch-Sulzberger13,14
Se trata de un TNC complejo, infrecuente y hereditario ligado al cromosoma Xq28. Lo sufren exclusivamente las mujeres, y es letal en los varones.
Alteraciones cutáneas
Las manifestaciones cutáneas son evolutivamente diferentes y a veces solapadas. En una primera fase se observan lesiones eritematosas, vesiculares o bullosas; éstas están presentes al nacimiento o en los primeros días de vida, aunque pueden durar varios meses. Se sucede una segunda fase con lesiones verrugosas, pustulares o queratósicas; estas nuevas alteraciones aparecen de novo o asientan sobre las primeras. Tienden a aparecer en los dos primeros meses de vida y se mantienen en ocasiones durante la lactancia. Estas lesiones pueden desaparecer en un tercer estadio o asociarse con una pigmentación cutánea anormal en forma de lesiones hiperpigmentadas pardas o grisáceas que adquieren formas peculiares (espirales, estrellas, aspecto de mármol). Estas alteraciones de la pigmentación se mantienen generalmente durante toda la infancia.
Otra manifestación cutánea relevante es la presencia de una alopecia cicatrizal que puede observarse hasta en el 30% de los pacientes.
Alteraciones neurológicas
Las manifestaciones centrales condicionan el pronóstico. Aparecen en el 30-50% de los niños. Las más prevalentes son el retraso mental, las parálisis espásticas, la ataxia cerebelosa y las crisis convulsivas (10%).
En la neurorradiología, el hallazgo más frecuente es la atrofia generalizada, las alteraciones de la sustancia blanca subcortical y las alteraciones cerebelosas.
Otras alteraciones
Incluyen trastornos oculares diversos (anomalías de la retina o vítreo, fibroplasia retrolenticular, malformaciones) que se pueden objetivar hasta en el 30% de los casos.
Las deformidades esqueléticas o faciales, así como las alteraciones congénitas renales o cardíacas también han sido descritas.
Diagnóstico
El diagnóstico es predominantemente clínico, y se debe realizar un adecuado diagnóstico diferencial con otros trastornos de la pigmentación cutánea.
Melanosis neurocutánea15,16
Es un síndrome infrecuente, generalmente esporádico, que afecta a ambos sexos por igual.
Alteraciones cutáneas
Se caracteriza por la presencia de nevos gigantes, pilosos, observables al nacimiento y localizados en cualquier parte del cuerpo, especialmente en abdomen y espalda. Son variables en número y tamaño (fig. 6). La tercera parte de los pacientes con nevo gigante desarrollan afección neurológica, con mayor frecuencia si los nevos asientan en cuero cabelludo o cara. Hasta un 10% de las lesiones maligniza.
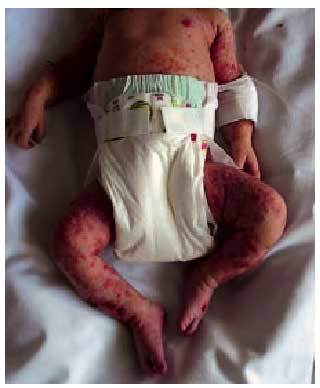
Fig. 6. Incontinentia pigmenti en recién nacido.
Alteraciones neurológicas
El melanoma leptomeníngeo es la manifestación más específica de este TNC. Estos melanomas son observables generalmente en la primera infancia. Aunque el estudio del líquido cefalorraquídeo puede ser útil para el cribado inicial, la RM es la técnica de elección para
el diagnóstico de las lesiones meníngeas.
Clínicamente puede cursar de forma asintomática, aunque más de la mitad de los casos asociará hidrocefalia por invasión de los espacios subaracnoideos. El pronóstico de las formas sintomáticas es habitualmente malo con independencia de los tratamientos empleados.
Se han descrito casos con retraso mental, crisis, déficit motores y otros.
Diagnóstico
Aunque los criterios diagnósticos se mantienen en debate, esencialmente se basan en la presencia de grandes o numerosos nevos cutáneos pigmentados, asociados a melanosis o melanoma meníngeo.
Síndrome de Sturge-Weber17,18
El síndrome de Sturge-Weber o angiomatosis encefalotrigeminal es un TNC poco frecuente y generalmente esporádico, aunque se han referido patrones de herencia autosómico dominante y recesivo.
Alteraciones cutáneas
El angioma facial o naevus flammeus es la manifestación dermatológica que caracteriza esta enfermedad. Es un nevo plano, rojo violáceo que afecta predominantemente a la cara de forma uni o bilateral (30%), asociado en ocasiones a una hipertrofia ipsilateral de partes blandas.
Alteraciones neurológicas
Comprenden básicamente la presencia de angiomas leptomeníngeos que afectan a la piamadre. Subyacentemente, puede asociar calcificaciones corticosubcorticales.
Clínicamente se manifiesta por crisis epilépticas en el 75-90% de los pacientes, que aparecen en la mitad de los casos en el primer año de vida. Son parciales en su inicio. La presencia de crisis refractarias puede apuntar la necesidad de tratamiento neuroquirúrgico, mediante la resección de áreas afectadas o incluso la hemisferectomía completa.
El retraso mental se objetiva en el 40% de los casos y tan sólo afecta a pacientes que han presentado convulsiones. Otro hallazgo habitual (30%) es la presencia de hemiparesia y hemianopsia.
Alteraciones oculares
Con carácter homolateral al angioma facial, en la tercera parte de los casos se observa un angioma ocular que afecta a la coroides y esclerótica, y que puede provocar glaucoma (30-50%), dolor orbitario, alteración de la agudeza visual y desprendimiento de retina en los primeros años de vida.
Diagnóstico
El diagnóstico se establece por la demostración del angioma pial en estudios neurorradiológicos y ante la presencia de angioma facial u ocular, aunque éstos pueden estar ausentes en el 13% de los pacientes.
La TC craneal y la radiografía de cráneo pueden revelar precozmente la presencia de las calcificaciones. La RM cerebral con contraste es la técnica de elección para el estudio del angioma leptomeníngeo. El electrocardiograma suele evidenciar una clara asimetría en la actividad de base.
Ataxia-telangiectasia19,20
La ataxia-telangiectasia es un TNC autosómico recesivo cuyo gen causante se ha localizado en el cromosoma 11 (11q22-33). Afecta por igual a ambos sexos, con una incidencia de 1/40.000 recién nacidos vivos.
Alteraciones oculocutáneas
Las telangiectasias se observan generalmente a partir de los 3 años en la conjuntiva bulbar. En la mitad de los casos se acompañan posteriormente por telangiectasias en párpados, pabellones auriculares, fosas poplíteas y regiones antecubitales.
Alteraciones neurológicas
La presencia de ataxia lentamente progresiva y de inicio precoz es una constante. Desde el punto de vista neurorradiológico se asocia con una atrofia cerebral y cerebelosa inespecífica. La evolución es mala; la mayor parte de los pacientes es incapaz de caminar a los 10-15 años.
Clínicamente la ataxia se acompaña por movimientos oculares anormales. En el 25% de los casos se asocia con coreoatetosis y, en la tercera parte de los pacientes, con retraso mental generalmente leve.
Alteraciones inmunológicas
Son frecuentes los defectos inmunológicos humorales y celulares. Los recuentos linfocitarios son bajos, la respuesta de los tests dérmicos son pobres y la proliferación leucocitaria ante mitógenos es deficitaria. Los valores de IgA e IgE son bajos o están ausentes, los de IgG, normales o bajos, y los de IgM, normales o altos.
Estas alteraciones se manifiestan por infecciones frecuentes, especialmente respiratorias (50-75%), y comúnmente graves.
Otras alteraciones
Incluyen el retraso ponderoestatural y la aparición de tumores malignos, habitualmente linforreticulares. Esta última circunstancia se describe hasta en el 50% de los casos fallecidos.
Diagnóstico
Aunque el diagnóstico se establece de acuerdo a los hallazgos clinicoanalíticos referidos, la elevación de la alfafetoproteína y del antígeno carcinoembriogénico y la presencia de anomalías cromosómicas inconstantes (inversión y traslocación de los cromosomas 7 y 14) son marcadores precoces y fiables para el diagnóstico.
Enfermedad de Von Hippel-Lindau21
La enfermedad de Von Hippel-Lindau o angiomatosis retinocerebelosa es un TNC con un patrón de herencia autosómico dominante, con una penetrancia del 80-90%. El gen ha sido mapeado en el cromosoma 3p35-p26.
Alteraciones oculares
Las manifestaciones oculares se observan a partir de la segunda década de la vida. Se caracterizan por la presencia de angiomas retinianos, generalmente múltiples y con frecuencias bilaterales. Pueden cursar con hemorragias, desprendimiento de retina, glaucoma o cataratas.
Alteraciones neurológicas
Comprenden los hemangioblastomas cerebelosos y medulares fácilmente objetivables en los estudios neurorradiológicos. Los primeros se observan en la mitad de los pacientes y se manifiestan clínicamente por un síndrome hipertensivo intracraneal o un síndrome cerebeloso completo. Los segundos son habitualmente asintomáticos.
Otras alteraciones
En este apartado se incluyen la policitemia (20%), los quistes renales o pancreáticos (30%), el carcinoma renal (40%) y el feocromocitoma (10-15%).

Diagnóstico
Se establece de acuerdo a los hallazgos descritos. La vigilancia debe extremarse ante la presencia de una lesión compatible en retina o cerebelo. Aproximadamente el 20% de los pacientes con angiomas de retina desarrollará complicaciones neurológicas, y en contrapartida, el 45% de los pacientes con hemangioblastomas cerebelosos tiene afección retiniana. De igual forma, son necesarios los controles clínicos, ecográficos y analíticos específicos para el cribado periódico de las complicaciones abdominales.
Síndrome de Klippel-Treaunay22
Es un síndrome infrecuente y esporádico, reconocible al nacimiento.
Alteraciones cutáneas
Las manifestaciones cutáneas incluyen los angiomas capilares y los hemangiomas cavernosos de trayectos irregulares. Pueden asociarse con trastornos de la pigmentación. La hipertrofia corporal, especialmente de extremidades inferiores, es habitual y no siempre ipsilateral a las lesiones de piel.
Alteraciones neurológicas
La macrocefalia es común. Cuando las lesiones cutáneas afectan a la cara o la calota, los pacientes pueden asociar retraso mental.
La neuroimagen pueden poner de manifiesto la presencia de malformaciones arteriovenosas, drenajes venosos anómalos, calcificaciones intracraneales o trastornos de la migración neuronal.

Diagnóstico
El diagnóstico se basa en los hallazgos clínicos; la RM es la técnica de elección para el estudio de las lesiones en piel y SNC.
Conclusiones
A través de estas breves anotaciones hemos tratado de resumir los TNC más frecuentes. La descripción detallada de la totalidad de las alteraciones cutáneas con significación neurológica supondría la revisión de un millar de enfermedades que sobrepasan las dimensiones de cualquier artículo23.
El propósito inicial y final de esta revisión es esencialmente alertar al pediatra sobre la relación entre las manifestaciones cutáneas y la clínica neurológica. Aunque históricamente este propósito se ha ido consiguiendo de forma inconstante en la neurofibromatosis, nuestra experiencia nos ha demostrado la falta de información sobre otros TNC, cuya prevalencia y gravedad obligan al especialista de atención primaria a su reconocimiento precoz.
