








La policondritis recidivante (PR) es una enfermedad inflamatoria sistémica y episódica de etiología desconocida y base autoinmune, caracterizada por lesiones inflamatorias recidivantes y potencialmente destructivas, que afecta a las estructuras cartilaginosas, el sistema cardiovascular y los órganos de los sentidos.
Fue descrita inicialmente por Jaksch-Wartenhorst en 1923 con el nombre de policondropatía. Posteriormente, otros autores publicaron nuevos casos y utilizaron otras denominaciones, como policondritis crónica atrófica, siendo la propuesta por Pearson (policondritis recidivante) la más ampliamente aceptada.
La distribución según sexos varía en las diferentes series desde una afección por igual en ambos hasta un ligero predominio en la mujer (3:1). Suele aparecer entre los 30 y los 60 años, con un pico de incidencia en la quinta década de la vida. Aunque es una afección que puede presentarse de forma aislada, se asocia con relativa frecuencia a otras enfermedades autoinmunes sistémicas, patologías hematológicas o enfermedades endocrinas. En un tercio de los casos la PR se asocia a artropatías inflamatorias y a otras enfermedades autoinmunes (tabla 1).
Etiopatogenia
La etiología de la PR es desconocida, aunque sus características indican que los mecanismos autoinmunes son importantes en su desarrollo, dado el hallazgo de una respuesta inmune, tanto humoral como celular, contra diferentes componentes del cartílago. La presencia de anticuerpos anticolágeno tipo II durante el episodio agudo explicaría el desarrollo de una respuesta autoinmune contra epítopes de algún componente del cartílago, con aparición posterior de un infiltrado inflamatorio pericondral. Lo mismo ocurriría en el resto de los tejidos a través de epítopes compartidos. La inmunofluorescencia del cartílago afectado demuestra depósitos de IgG, IgA, IgM y C3, lo que indica la existencia de una lesión mediada por inmunocomplejos. Desde un punto de vista inmunogenético, se han realizado pocos estudios para caracterizar la PR, aunque recientemente se ha detectado un aumento de la incidencia de HLA-DR4 sin subtipo predominante en estos pacientes. Experimentalmente se ha observado la aparicion de condritis en ratas inmunizadas con colágeno tipo II.
Manifestaciones clínicas
Como cualquier otra enfermedad autoinmune sistémica, la PR puede afectar a numerosos órganos, con una especial predilección por la afección condral y articular (fig. 1).
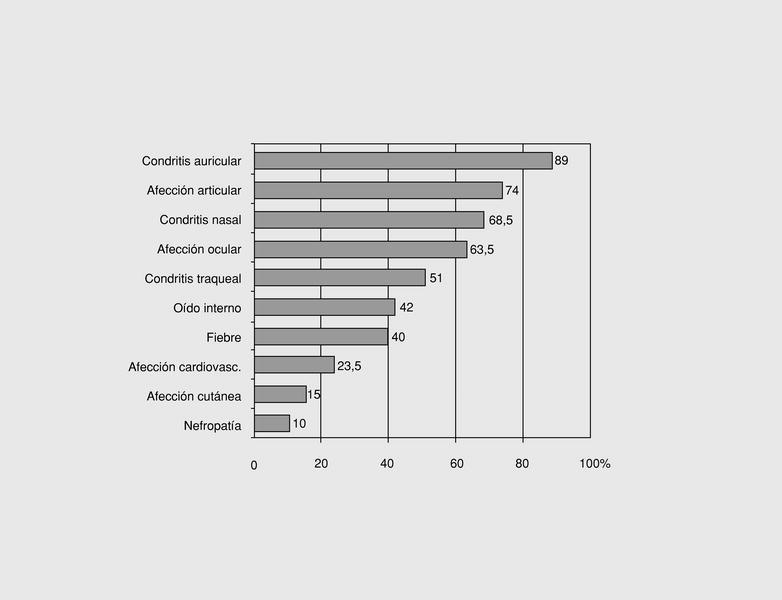
Fig. 1. Frecuencia de las principales manifestaciones clínicas de la policondritis recidivante.
Manifestaciones generales
El cuadro clínico puede iniciarse con fiebre, pérdida de peso y afección del estado general.
Condritis
Condritis auricular
Es la afección más frecuente, bien de forma localizada o difusa, uni o bilateralmente (fig. 2). Su instauración es rápida y a menudo es la forma de presentación de la enfermedad. Puede existir otorrea serosa con lesión permanente y reblandecimiento del cartílago auricular, dando lugar a la típica oreja de cocker.
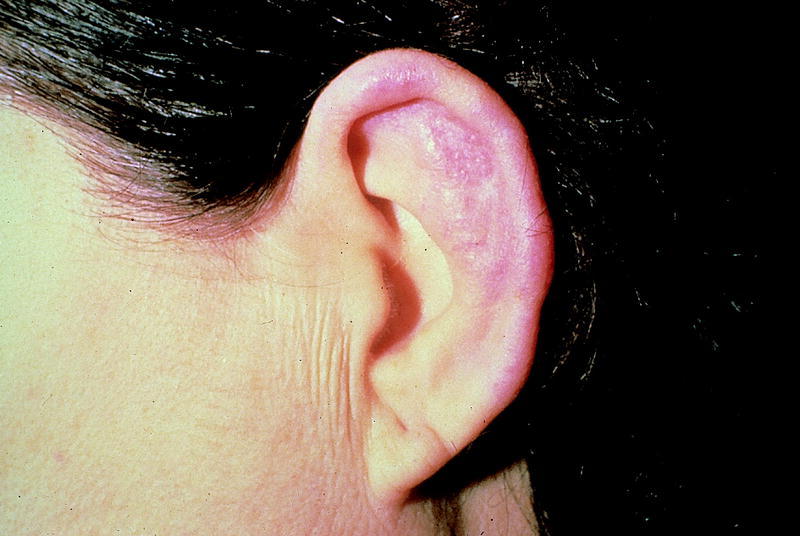
Fig. 2. Condritis del pabellón auricular.
Condritis nasal
Puede aparecer al inicio de la enfermedad o bien a lo largo de su evolución. El cuadro agudo puede pasar inadvertido, caracterizándose por discreto dolor y ocasionalmente rinorrea y/o epitaxis. La condritis recidivante de los cartílagos de la pirámide nasal produce una deformidad característica en silla de montar.
Condritis laríngea y traqueobronquial
La condritis laríngea provoca afonía completa que simula una laringitis banal. El paciente puede presentar estridor, sibilancias y tos, con disnea respiratoria asmatiforme. La inflamación de la tráquea y del cartílago tiroides puede ocasionar dolor y evolucionar a esteno sis local o difusa. Estas manifestaciones son más frecuentes en mujeres y son la primera causa de muerte. Además, este tipo de afección predispone a sobreinfecciones.
Condritis costales
Son menos frecuentes y poco relevantes. Los pacientes presentan dolor con discreta tumefacción de las uniones condrocostales. Si la afección es importante, pueden dar lugar a la deformación en pectus excavatum.
Artritis
Las manifestaciones articulares son muy frecuentes durante el curso de la enfermedad. Se pueden presentar como una poliartritis o bien como una oligoartritis aguda o subaguda. La afección articular suele ser intermitente y asimétrica, no deformante ni erosiva, localizada en pequeñas y grandes articulaciones, que se resuelve de forma espontánea o con antiinflamatorios no esteroides.
Manifestaciones oculares
Epiescleritis y escleritis son las manifestaciones oculares más frecuentes. También puede encontrarse conjuntivitis, infiltrado corneal periférico, iridociclitis, desprendimiento de retina y neuropatía isquémica del nervio óptico.
Alteraciones audiovestibulares
La hipoacusia puede presentarse por estenosis del conducto auditivo externo secundaria a inflamación, así como también por otitis media y obstrucción de las trompas de Eustaquio por condritis del segmento nasofaríngeo. También pueden observarse trastornos neurosensoriales cocleares o vestibulares, posiblemente por vasculitis de la arteria auditiva interna o de sus ramas vestibular o coclear.
Trastornos cardiovasculares
Aunque son poco frecuentes, constituyen la segunda causa de muerte en los pacientes con PR. La insuficiencia aórtica es la valvulopatía más frecuente, y se debe a una dilatación progresiva del anillo aórtico y de la aorta ascendente, o bien a la destrucción de las sigmoideas aórticas. Rara vez se presenta al inicio de la enfermedad y es más frecuente en el sexo masculino. La insuficiencia mitral y tricuspídea son muy raras, así como la pericarditis y la miocarditis. Puede existir afección de vasos de gran y medio calibre sin lesión cardíaca, tales como aneurismas de arteria subclavia y de aorta torácica o abdominal, y fenómenos de trombosis arterial, sobre todo de mediano y gran calibre.
Alteraciones cutaneomucosas
Se presentan en el 30% de los pacientes, y puede observarse cualquier tipo de lesión cutánea (pápulas, nódulos, vesículas o ampollas). En la biopsia cutánea se puede observar vasculitis, en general leucocitoclástica, aunque también puede ser de tipo granulomatoso o necrosante.
Alteraciones renales
La afección renal en la PR es poco frecuente. La alteración más común es la glomerulonefritis mesangial, seguida de la glomerulonefritis necrosante focal y segmentaria. La inmunofluorescencia revela depósitos de C3, IgG e IgM en el mesangio.
Alteraciones del sistema nervioso central
Son poco frecuentes y consisten en lesiones de los pares craneales II, VI, VII y VIII, signos cerebelosos, convulsiones y demencia.
Diagnóstico
Laboratorio general
Las alteraciones del laboratorio son inespecíficas aunque útiles, principalmente para excluir otros procesos patológicos y para el seguimiento de la actividad de la enfermedad. El parámetro más útil para monitorizarla es la velocidad de eritrosedimentación, la cual se encuentra elevada en el 90% de los casos durante la actividad de la enfermedad. La anemia está presente en las dos terceras partes de los pacientes, y posee las mismas características de la anemia asociada a los procesos crónicos. Finalmente, otros hallazgos de laboratorio son leucocitosis, trombocitosis, elevación de alfa 2 o betaglobulinas e hipergammaglobulinemia policlonal.
Inmunología
Los exámenes inmunológicos no contribuyen al diagnóstico. Los anticuerpos antinucleares generalmente son negativos. Se ha descrito la presencia de ANCA (generalmente p-ANCA) en un bajo porcentaje de pacientes, así como de anticuerpos antifosfolipídicos y factor reumatoide.
En un 20% de los pacientes se pueden encontrar anticuerpos anticolágeno tipo II, pero su frecuencia es demasiado baja para que puedan utilizarse como un marcador diagnóstico.
Anatomía patológica
No existen hallazgos anatomopatológicos patognomónicos. En la fase aguda se observa un infiltrado polimorfonuclear inflamatorio focal en las estructuras pericartilaginosas, con pérdida de los proteoglicanos de la matriz. Posteriormente se observa degeneración de condrocitos con invasión de la matriz cartilaginosa y reemplazo del cartílago por tejido de granulación. En fases avanzadas de la enfermedad podemos encontrar fibrosis o calcificación.
La biopsia raramente es necesaria para el diagnóstico, salvo cuando sea precisa para realizar un diagnósti co diferencial con la enfermedad de Wegener o el granuloma de la línea media.
Criterios diagnósticos
Mac Adam et al propusieron en 1976, a partir del estudio de 23 pacientes, una serie de 6 criterios clínicos (condritis auricular bilateral, poliartritis seronegativa inflamatoria no erosiva, condritis nasal, inflamación ocular, condritis del tracto respiratorio y alteraciones vestibulares y/o cocleares), exigiendo para el diagnóstico de PR la presencia de tres de ellos más una confirmación histológica (tabla 2). Posteriormente, y a fin de evitar la realización sistemática de biopsias, Mitchet et al dividieron los criterios anteriores en mayores y menores (tabla 3).




Aunque ninguna de las dos clasificaciones ha sido universalmente aceptada, la propuesta por Mitchet et al se considera la más práctica, ya que evita la realización de biopsias, que raramente son necesarias para el diagnóstico de la enfermedad.
Diagnóstico diferencial
En el proceso diagnóstico de la PR se deben considerar siempre una serie de enfermedades que pueden asemejarse a ésta. Las patologías que presentan mayores inconvenientes a la hora de realizar un diagnóstico diferencial son los procesos infecciosos, las vasculitis sistémicas o localizadas y otras enfermedades autoinmunes sistémicas. Las infecciones agudas y crónicas del pabellón auricular deben distinguirse de la PR. La bilateralidad del proceso y su carácter recidivante orientarán el cuadro hacia el diagnóstico de PR.
La granulomatosis de Wegener es la entidad que más dificultad puede originar en el diagnóstico diferencial, ya que ambas patologías comparten manifestaciones clínicas, como la afección de las vías aéreas superiores y del tabique nasal. La condritis auricular, tan característica en la PR, es excepcional en el Wegener, mientras que en este último se observa con frecuencia la afección pulmonar y renal, rara en la PR.
El síndrome de Cogan es otra entidad a tener en cuenta, ya que puede presentar síntomas vestibulococleares y queratitis intersticial.
En todos estos casos, la biopsia y los cultivos apropiados serán de mucha utilidad para la definición diag nóstica.
Pronóstico
El curso clínico de la PR es muy variable: desde grave y rápidamente progresivo hasta más benigno, con brotes esporádicos de la enfermedad. De todas formas, en la mayoría de los casos el cuadro evoluciona de manera episódica, encontrando una minoría que presenta sintomatología continua.
La supervivencia de estos pacientes es inferior a la de la población general, siendo a los 5 años del 75% y a los 10 años del 55%. Las causas de muerte más frecuentes son las infecciones respiratorias (neumonía), seguidas de la insuficiencia respiratoria por obstrucción laringotraqueal, la disección o rotura de aneurismas, las valvulopatías y las vasculitis. El inicio de la enfermedad en pacientes menores de 50 años y la presencia de anemia, deformidad nasal, artritis, estenosis laringotraqueal, vasculitis y microhematuria podrían considerarse como factores predictivos de mal pronóstico.
En la serie de Trentham y Le, se observaron como principales complicaciones de la enfermedad el colapso traqueal, las valvulopatías graves y las secuelas oftalmo lógicas, mientras que la mortalidad en dicha serie fue del 6%.
Tratamiento
Antiinflamatorios no esteroides (AINE)
El tratamiento de determinadas manifestaciones, tales como condritis auricular, nasal, artritis y fiebre, se basa en la utilización inicial de AINE. Habitualmente este tratamiento no suele ser totalmente efectivo, y se requiere la administración de esteroides por vía oral.
Corticoides
Se usan en la gran mayoría de las manifestaciones clínicas y a dosis que oscilan entre 20 y 60 mg/día hasta controlar la sintomatología. En algunos casos concretos en que a dosis de 1 mg/kg/día no se ha obtenido respuesta terapéutica, o ante una situación de riesgo vital, se pueden utilizar bolos de 1 g de metilprednisolona intravenosa durante 3 días consecutivos, con resultados favorables.
La disminución de los corticoides por debajo de una determinada dosis desencadena, a veces, la reaparición de la sintomatología; en tales casos se requieren inmunodepresores como ahorradores de esteroides. Éstos también están indicados en los casos de fracaso terapéutico con el tratamiento de primera línea.
Inmunodepresores
El fármaco de segunda línea más eficaz y con menores efectos adversos es el metotrexato, a dosis de 17,5 mg/ semana, cuya adición al tratamiento corticoide suele conseguir, según diversas series, un requerimiento de esteroides de tan sólo 5 mg/día. Otros inmunodepresores utilizados son la azatioprina a dosis de 2 a 3 mg/kg /día, y la ciclofosfamida vía oral o en bolos intrave nosos, esta última especialmente en casos graves de estenosis u obstrucción de las vías aéreas superiores, glomerulonefritis rápidamente progresiva y manifestaciones vasculíticas graves. También se ha empleado la ciclosporina, a dosis de 3 a 5 mg/kg/día.
Otros fármacos
La colchicina, a dosis de 1 mg/día, se puede utilizar para controlar la condritis y los síntomas cutáneos menores. Los resultados del tratamiento con D-penicilamina, sulfasalazina y minociclina son inciertos. Finalmente, se ha comunicado el efecto benéfico de la terapia con anticuerpos monoclonales anti-CD4 en dos pacientes con PR.
Tratamiento quirúrgico
En casos concretos de estenosis o colapso de la vía aérea está indicada la cirugía torácica reparadora. También pueden ser motivo de tratamiento quirúrgico las valvulopatías graves y los aneurismas aórticos. Por último, las intervenciones quirúrgicas correctoras, como las de las deformidades nasales mediante prótesis, deberán realizarse cuando la actividad inflamatoria esté controlada.
Bibliografía recomendada
Eng J, Sabanathan S. Airway complications in relapsing polychondritis. Ann Thorac Surg, 1991; 51: 686-692.
Firestein GS, Gruber HE. Mouth and genital ulcers with inflamed cartilagr: MAGIC Syndrome. Am J Med 1985; 79: 75-62.
Foirdart JM, Abe S. Antibodies to type II collagen in relapsing polychondritis. N Engl J Med 1978; 299: 1203-1207.
Lang B, Rothenfussser A. Susceptibility to relapsing polychondritis is associated with HLA DR4. Arthritis Rheum 1993; 36: 660-664.
McAdam LP, O'Hanlan MA. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine (Baltimore) 1976; 55: 193-215.
Mitchet CJ, McKenna CH. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med 1986; 104: 74-78.
Trentham DE, Le CH. Relapsing polychondritis. Ann Intern Med 1998; 129: 114-121.
Vinceneux P, Piette JC. Polychondrite atrophiante. En Kahn MF, Piette JC, editores. Maladies et syndromes systémiques (4.a ed.). Médicine Scien., 2000: 623-649.