







Las alteraciones hepáticas causadas por los fármacos constituyen la mayor parte de las hepatopatías secundarias a tóxicos y son frecuentes en la población. Algunos datos pueden darnos una idea más exacta de esta ase veración: cerca del 10% de las hepatopatías pueden
deberse a efectos secundarios de los medicamentos; de los casos de fracaso hepático fulminante un 15-30% se deben a fármacos; un 2-3% de las admisiones hos pitalarias por reacciones adversas a medicamentos se producen por los efectos secundarios que tienen sobre el hígado; el 5% de las hospitalizaciones por ictericia están motivadas por hepatotoxicidad farmacológi ca, etc.
Muy probablemente la realidad supere esos datos, puesto que gran parte de estas lesiones hepáticas pasan desapercibidas o no son declaradas debidamente a los organismos responsables de su registro. Además, es un hecho que con el transcurso del tiempo está aumentando su incidencia, lo que puede deberse tanto a la introducción de nuevas moléculas por parte de la industria farmacéutica como al reconocimiento tardío del efecto tóxico de algunos fármacos que estaban comercializados con anterioridad.
Como muy bien se apuntaba en un editorial relativamente reciente de esta revista, la problemática de la hepatotoxicidad por fármacos se puede definir en dos vocablos: amplitud y complejidad. Su amplitud se entiende por el elevado número de fármacos que tenemos a nuestra disposición, así como por el hecho de que cualquier hepatopatía puede estar causada por ellos. En cuanto a la complejidad, es la susceptibilidad individual a padecer estas lesiones uno de los aspectos involucrados, del cual nos ocuparemos en los siguientes epígrafes.
Metabolismo hepático de los fármacos
Es bien conocido que la mayoría de los medicamentos son compuestos liposolubles (no polares). Mediante el proceso de biotransformación que sufren en el hígado se pueden convertir en sustancias químicas hidrosolubles (polares), las cuales pueden fácilmente ser eliminadas hacia la bilis o, por medio del plasma, hacia la orina.
El metabolismo hepático de un elemento liposoluble comprende dos fases denominadas fase I y fase II (fig. 1). En la fase I, las hidrolasas y oxidorreductasas, que son enzimas situadas en los microsomas del retículo endoplasmático liso de los hepatocitos, producen los cambios necesarios, generalmente la adición de un grupo hidroxilo, para que cualquier sustancia lipofílica se transforme en una sustancia más hidrófila. La reacción enzimática más importante en esta fase es la oxidación, y se efectúa mediante la acción de un sistema enzimático denominado de la monoaminooxidasa. Éste consta de tres elementos: una hemoproteína (citocromo P-450), una flavoproteína (citocromo-NADPH-c reductasa) y un lípido (fosfatidilcolina). Entre ellos, el más destacado es el citocromo P-450, que actúa como aceptor de electrones de una gran variedad de reacciones de oxidación. Tras esta primera fase ocurre la segunda (fase II), que consiste en la conjugación de los compuestos polares producidos en la fase I, mediante otras enzimas (transferasas), situadas en los microsomas, citosol o mitocondrias, que son capaces de adicionar sustratos endógenos como el ácido glucurónico, el ácido sulfúrico, la glicina o el glutation a esas sustancias polares. La mayor parte de estas enzimas están situadas en las regiones periportales.
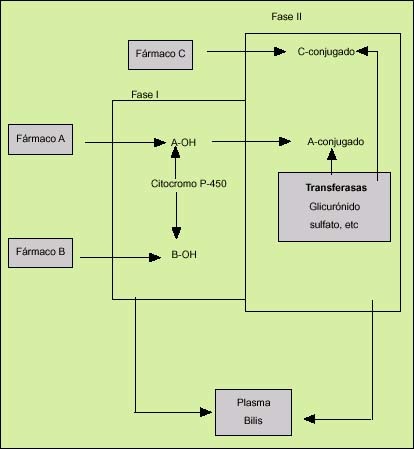
Fig. 1. Biotransformación de los fármacos en el hígado. El fármaco A puede biotransformarse por medio de las fases I y II. El B sólo mediante la fase I. El C es secretado desde el hepatocito tras la fase II.
Además de su intervención en la biotransformación, la segunda fase es primordial en muchas ocasiones para evitar la toxicidad de los metabolitos activos producidos por la acción de las enzimas de la fase I, aunque en ocasiones los compuestos conjugados pueden ser más tóxicos que el metabolito activo o que el propio producto orgánico inicial.
Factores asociados con la hepatotoxicidad farmacológica
Hay dos factores primordiales. Unos son dependientes del propio fármaco y los otros están relacionados con el paciente. Estos últimos son más frecuentes e importantes.
Factores relacionados con el fármaco
No sólo influye la composición química del medicamento, también es importante su estructura molecular. Así, por ejemplo, el ácido tienílico no tiene una hepatotoxicidad directa y puede desencadenar, mediante un mecanismo inmunoalérgico, una hepatitis que remeda a la hepatitis autoinmune con positividad de los anticuerpos anti-LKM tipo 2; sin embargo, un enantiómero del mismo sí que tiene un mecanismo directo de hepatotoxicidad y no genera la hepatopatía inmunoalérgica.
Factores relacionados con el paciente
Edad
Al igual que sucede en otras reacciones adversas por fármacos, la hepatotoxicidad causada por medicamentos es mucho más frecuente en los ancianos. Este hecho se debe a múltiples circunstancias, entre las cuales se puede citar la polimedicación, la coexistencia de otras enfermedades y las alteraciones del metabolismo farmacológico (sistémicas y hepáticas) que se dan en los pacientes de edad avanzada. En lo que concierne a las alteraciones sistémicas, podemos subrayar aquí que algunos medicamentos lipofílicos sufren un cambio en el volumen de distribución con arreglo a la edad (p. ej., benzodiazepinas); en otros fármacos se modifica tanto su unión a proteínas como su eliminación renal. Además, en lo que respecta a las alteraciones hepáticas, los cambios fisiológicos del hígado tras el paso de los años (menor masa hepatocitaria total, disminución en el flujo sanguíneo hepático, etc.) pueden ser importantes para incrementar la susceptibilidad hepática a las lesiones hepáticas por fármacos. No olvidemos que en los ancianos puede ser menor la síntesis de los factores protectores de la hepatotoxicidad, lo que puede justificar una disminución de vitaminas antioxidantes, glutation, etc. También, en estas edades, las modificaciones de las respuestas inmunológicas pueden condicionar una mayor facilidad en estas personas para padecer lesiones hepáticas.
En este colectivo no sólo es mayor el riesgo de hepatotoxicidad, sino que además la gravedad de las lesiones hepáticas suele ser mayor, y como consecuencia existe una mayor mortalidad y, por lo tanto, un peor pronóstico en estos pacientes. Varios factores pueden estar involucrados en ello, como una menor respuesta regenerativa hepática, la afección renal, los déficit nutricionales, etc.
Con todo, resulta muy difícil responder a qué edad empieza a incrementarse el riesgo de la hepatotoxicidad por fármacos. En algunos medicamentos, como la isoniazida, el halotano o la clorpromazina, este límite puede estar entre los 30 y 40 años.
Por contra, en los niños es excepcional la existencia de estas reacciones adversas, con la excepción de la hepatotoxicidad por el ácido valproico (que es más frecuente en menores de 3 años) y la causada por los salicilatos. Ambas lesiones pueden ser el resultado de una mayor susceptibilidad a la lesión mitocondrial en la infancia.
Sexo
Algunos medicamentos causan con frecuencia lesiones hepáticas en la mujer (tabla 1). Por el contrario, otros fármacos, uno de cuyos ejemplos puede ser la amoxicilina-ácido clavulánico, originan más reacciones hepatotóxicas en el varón. Se desconoce cuáles son los factores que predisponen a esta diferencia según el sexo, aunque teóricamente podría deberse a un efecto adverso de los estrógenos y/o un efecto protector de los andrógenos.

Hormonas
Mediante su actuación sobre el sistema oxidativo microsomal enzimático (MEOS) también conocido como oxidasa de función mixta o de la monooxigenasa, el hipertiroidismo aumenta la susceptibilidad a las lesiones hepáticas causadas por fármacos, mientras que el hipotiroidismo protege de esas agresiones. Además de la acción directa de las hormonas tiroideas, en estos efectos también pueden estar involucradas indirectamente las catecolaminas. Es lógico, por tanto, que el propiltiouracilo (fármaco antitiroideo) ejerza un efecto protector sobre la hepatotoxicidad del paracetamol, el tetracloruro de carbono, los salicilatos y el alcohol. Este fármaco, además de tener actividad antitiroidea, actúa como un subrogado del glutation (sustancia antioxi dante).
El efecto experimental de los glucocorticoides es el de incrementar la toxicidad hepática del tetracloruro de carbono, mientras que la adrenalectomía protege de esta lesión.
La diabetes mellitus en los animales de experimentación potencia la hepatotoxicidad de algunas substancias como la galactosamina y la tioacetamida, lo cual parece depender de la inducción del citocromo P-450 2E1 por las cetonas. En el ser humano, salvo en el caso del antituberculoso etionamida y del metotrexato, la existencia de diabetes no incrementa el riesgo de hepatotoxicidad. Ello se debe a que el efecto inductor del citocromo antes mencionado se ve compensado por una mayor glucuronización que existe en esta enfermedad endocrinológica. También es conveniente conocer que la administración de glucosa tiende a generar un mayor riesgo de hepatotoxicidad, debido a que disminuye la respuesta regenerativa hepática secundaria a una lesión sobre el hepatocito.
La presencia de obesidad potencia el riesgo de lesiones hepáticas causadas por algunos fármacos (tabla 2). Este mayor riesgo depende de dos causas. La primera, el mayor componente lipídico de estos pacientes; la otra, la inducción enzimática del citocromo P-450 2E1 generada por la propia obesidad.

Factores nutricionales
El estado nutricional puede modificar desde el punto de vista teórico el riesgo de la hepatotoxicidad causada por fármacos por varios motivos. Entre otros una modificación de las enzimas que intervienen en la fase I (citocromos P-450) o en la fase II del metabolismo hepático de los fármacos. Además, de modo indirecto, por la mayor susceptibilidad lesional causada por la metamorfosis grasa que acompaña a algunas deficiencias nutricionales (p. ej., déficit proteico).
La deprivación de proteínas disminuye la cantidad y la actividad de los citocromos P-450, circunstancia que explica que disminuya el riesgo de hepatotoxicidad causada por algunas sustancias (p. ej., tetracloruro de carbono y dimetilnitrosaminas).
Otros efectos nutricionales sobre la familia de los citocromos P-450 pueden observarse en la tabla 3.

El ayuno condiciona un incremento del riesgo de hepatotoxicidad del paracetamol y de otros elementos, como la tioacetamida y el bromobenzeno. Esta alteración puede deberse a la disminución de las reservas del glucógeno hepático y de la glucuronización, así como a la existencia de unas menores reservas de glutation hepático en estos casos.
El consumo de algunos vegetales, como ocurre dentro del grupo de las crucíferas, como la coliflor y las coles de Bruselas, puede ser capaz de inducir el sistema de los citocromos P-450 mediante la acción de los indoles contenidos en estos alimentos, y de esta forma se puede potenciar la hepatotoxicidad de algunos fármacos como el paracetamol.
Hepatopatías previas
Aunque por tradición clínica se suponía que el riesgo de hepatotoxicidad era más acusado en los pacientes con una hepatopatía previa, no existen en la actualidad evidencias de peso que justifiquen una mayor susceptibilidad a las lesiones hepáticas causadas por fármacos en los pacientes con una hepatitis, una cirrosis o un hepatoma. La única excepción a este aserto puede ser la isoniazida en el caso de tener el paciente una hepatitis crónica.
Consumo de etanol
La ingestión crónica de este tóxico conlleva una modificación importante en la biotransformación hepática de algunos fármacos, lo cual puede redundar en un mayor riesgo de hepatotoxicidad farmacológica (tabla 4). Son múltiples los mecanismos que conducen a este incremento del riesgo. Entre otros, debemos mencionar la inducción enzimática ejercida por el etanol sobre algunos citocromos, como el citocromo P-450 2E1 y el P-450 3A, y la reducción de la síntesis de glutation hepático. Ambos fenómenos originan, por una parte, una mayor formación de metabolitos tóxicos por la inducción de los citocromos y, por otra, una menor posibilidad de eliminarlos por existir menos glu tation.

De todos los fármacos, el paracetamol es el paradigma de esta interrelación. En efecto, en los pacientes con consumo excesivo de alcohol existe un riesgo muy acentuado de hepatotoxicidad por el paracetamol, incluso con dosis consideradas dentro del rango terapéutico habitual, por lo que se aconseja que estos enfermos no tomen más de 2 g al día de este analgésico.
Interacciones con virus
Las infecciones virales pueden incrementar el riesgo de hepatotoxicidad por medicamentos. En el caso del virus de la hepatitis B no está descrita ninguna repercusión nociva en la clínica; no obstante, sí que se han identificado alteraciones del metabolismo oxidativo de algunos fármacos desde el punto de vista experimental.
La infección por el virus de la hepatitis C puede aumentar la susceptibilidad de los pacientes con tuberculosis a los efectos secundarios de los fármacos antituberculosos, y se ha llegado a recomendar por algún autor, con la finalidad de impedir estos efectos hepa totóxicos, que se intente antes del tratamiento antitu berculoso la eliminación de este virus hepatotropo mediante el uso de los antivirales habituales (interferón y ribavirina).
La infección por el VIH altera el metabolismo de la cafeína y disminuye el aclaramiento hepático de algunos fármacos, como la clindamicina y el fluconazol. Además, aumenta el riesgo de hepatotoxicidad por el cotrimoxazol así como la probabilidad de hepatotoxicidad por paracetamol a dosis terapéuticas.
Los mecanismos implicados en estas situaciones po drían depender de varias circunstancias. En primer lugar, un efecto viral directo sobre el proceso de biotransformación de los fármacos en el hígado; en segundo lugar, el interferón puede tener en algunos casos un efecto inhibidor sobre el citocromo P-450; en tercer lugar, podría existir una mayor sensibilidad a los fármacos por parte de los hepatocitos infectados por los virus; finalmente, en el caso del VIH, las alteraciones inmunológicas pueden desempeñar un papel muy destacado.
Factores genéticos
Muy probablemente los factores genéticos son los más importantes a la hora de generar una hepatotoxicidad por xenobióticos.
Entre las especies animales existen diferencias en la fase I y en la fase II del metabolismo hepático de los fármacos, tanto en lo que concierne a los aspectos cualitativos como a los cuantitativos, que son explicables mediante estos condicionantes genéticos.
En el ser humano, mediante estudios efectuados en gemelos univitelinos se ha demostrado de forma fehaciente la importancia de estos factores. En efecto, existen diferencias entre ellos en el metabolismo de algunos fármacos (dicumarol, fenilbutazona, etc.) que se interpretaron como diferencias en la fase I entre ambos.
Además, es bien conocida la influencia genética en el metabolismo de otros medicamentos. Es el caso de la isoniazida, en la cual existen con respecto a su acetilación sujetos que son acetiladores lentos y rápidos, siendo semejante la frecuencia de estos grupos entre los sujetos de raza negra y blanca, mientras que el 90% de la población asiática pertenece a los acetiladores rápidos. También se han comprobado inluencias genéticas en la fenitoína (hidroxilación y en la fase II de su metabo lismo).
Las circunstancias genéticas pueden influir tanto en los mecanismos enzimáticos como en los no enzimáticos del metabolismo hepático de los medicamentos. Así ocurre con el polimorfismo genético de los diversos citocromos P-450, que es estudiado mediante diversos tests; por ejemplo, el de la debrisoqu ina que valora el citocromo P-450 2D6 encargado de la fase I del metabolismo intrahepático de la perhexilina.
Rara vez puede existir la deficiencia genética de algún elemento protector de la hepatotoxicidad. Entre ellos el más importante sería el déficit del glutation. El mecanismo de herencia en estos casos es mendeliana recesiva, teniendo los homocigotos unos valores intracelulares de esta sustancia de un 15% de lo normal. Además de existir una mayor propensión a la hepatotoxicidad en estos pacientes, esta deficiencia se manifiesta por hemólisis, acidosis y alteraciones de los leucocitos polimorfonucleares.
Otras evidencias avalan la relación de la hepatotoxicidad con los mecanismos genéticos. Tal es el caso de la asociación de diversas lesiones hepáticas de origen medicamentoso con los HLA, como se puede ver en la hepatotoxicidad por clorpromazina (un 80% de los pacientes tienen el HLA DR6) o por diclofenaco (el 70% de estos enfermos tienen el HLA A11).
Es de esperar en el próximo futuro que el uso de los estudios genéticos in vitro mediante técnicas de amplificación molecular, como la reacción en cadena de la polimerasa (PCR), pueda identificar a las personas susceptibles al efecto tóxico de determinados fármacos. En este sentido una rama de la farmacología la farmacogenética está avanzando de forma espectacular, hecho que nos hace albergar grandes esperanzas para que este tipo de efectos adversos pueda reducirse de forma importante en los próximos años.
Bibliografía general
Bass NM, Ockner RK. Drug-induced liver disease. En: Zakim D, Boyer ThD, editores. Hepatology. Filadelfia: WB Saunders 1996; 962-1017.
Bessone F, Tanno H. Hepatotoxicidad inducida por antiinflamatorios no esteroides. Gastroenterol Hepatol 2000; 23: 200-205.
Bruguera M. Lesiones hepáticas por fármacos. Medicine (ed. esp.) 2000; 8: 448-452.
Camargo R, Lucena MI, Andrade RJ. Epidemiología actual de las hepatopatías tóxicas. Rev Gastroenterol 1999; 1: 664-684.
Encinas Sotillos A. Diagnóstico y prevención de las hepatopatías causadas por medicamentos. Med Integral 1997; 29: 145-154.
Encinas Sotillos A. Fármacos e Hígado (I). Salud Rural 1997; 14: 55-70.
Encinas A, Bruguera M. La hepatotoxicidad por fármacos: la prevención como objetivo. Gastroenterol Hepatol 1998; 21: 97-99.
Pérez V. Medicamentos e hígado. En: Vilardell F, Rodés J, Malagelada JR, Moreno E, Pajares JM, Pérez Mota A et al, editores. Enfermedades digestivas. Madrid: Ediciones CEA, 1990; 2081-2101.
Sánchez Tapias JM. Hígado y fármacos. Med Integral 1997; 29: 131-132.
Zimmerman HJ, Maddrey WC. Toxic and drug-induced hepatitis. En: Schiff L, Schiff ER, editores. Diseases of the liver. Filadelfia: JB Lippincott Company, 1993; 707-783.
Zimmerman HJ. Vulnerability of the liver to toxic injury. En: Zimmerman HJ, editor. Hepatotoxicity (2.ª ed.). Filadelfia: Lippincot Williams & Wilkins, 1999; 41-60.