













La epilepsia es un trastorno crónico de origen plurietiológico caracterizado por la aparición reiterada de crisis. Éstas se originan por una descarga hipersíncrona, brusca y excesiva de un grupo de neuronas.
La epilepsia en la infancia va a estar influida clínica y etiológicamente por las características de un sistema nervioso en maduración. Paralelamente, la respuesta a la medicación, el metabolismo de la misma y, al fin, el pronóstico dependerán también de la edad del paciente.
En los primeros meses de vida predominan las crisis de origen orgánico, es decir, la de peor pronóstico. En la lactancia y el período preescolar, la mayoría de las crisis epilépticas se asociarán del mismo modo con lesiones estructurales cerebrales; sin embargo, a esta edad aparecen característicamente la convulsión febril, trastorno de buen pronóstico inicial, y los síndromes epilépticos, muy bien caracterizados y de mal pronóstico, como el síndrome de West y el síndrome de Lennox-Gastaut. A partir de esta edad, hasta la adolescencia, aparecen las epilepsia de origen criptogénico, con gran carga genética, mientras que los síndromes epilépticos sintomáticos se hacen menos frecuentes; son características de esta edad la epilepsia de ausencias y la epilepsia rolándica benigna. Desde los 10 años de edad en adelante, se observan las epilepsias generalizadas primarias, con o sin mioclonías, desaparecen las parciales benignas y las de ausencias, y aumentan las crisis parciales sintomáticas1,2.
Incidencia
La incidencia de la epilepsia, según diferentes estudios, se sitúa entre 17,3 y 136/100.000 habitantes por año. Hauser y Kurland señalaron una tasa de 48,7/ 100.000/año para las crisis recurrentes3. En España se estima una incidencia aproximada de 40/100.000 habitantes4.
Se estima que la frecuencia de trastornos paroxísticos en la infancia es del 15%. El 10% de los mismos serán diagnosticados de crisis epilépticas, y la mitad de los mismos, tras su reiteración, serán diagnosticados de epilepsia.
La incidencia específica según la edad es más elevada en la primera década de la vida, especialmente durante el primer año, donde la incidencia llega a alcanzar cifras de 120-150/100.000 niños. La incidencia específica según el sexo refleja un claro predominio masculino en la mayor parte de los estudios.
La prevalencia de la epilepsia se sitúa, según los diversos estudios, entre 4 y 13 por 1.000 niños, habiendo excluido las convulsiones febriles. Hauser y Kurland señalaron una prevalencia de 0,37/100, elevándose hasta 0,58/100 en la primera década de la vida y hasta 0,63 en la segunda década3.
Extrapolando todos estos datos a la población española, en nuestro país habría más de 200.000 epilépticos. Igualmente, podríamos afirmar que cada pediatra controlará más de 10 pacientes epilépticos, y un porcentaje similar de pacientes que, de forma adicional, sufrirán una crisis epiléptica única.
Papel del pediatra ante el paciente epiléptico
Como apuntábamos en la introducción, el papel del pediatra en la atención del paciente pediátrico es esencial.
Podemos delimitar con carácter general las funciones que debería cumplir el mismo en el control de estos pacientes: control del paciente de riesgo neurológico; diagnóstico clínico de la primera crisis epiléptica (diagnóstico diferencial de otros trastornos paroxísticos);
conocimiento básico de la farmacología de los antiepilépticos, especialmente de sus efectos colaterales e interacciones; controles periódicos clínicos y analíticos;
recomendaciones generales para la vida diaria del paciente epiléptico, y actuación ante situaciones espe-
ciales.
Control del paciente de riesgo neurológico
Los factores de riesgo neurológico están estrechamente relacionados con la etiología de la epilepsia. La clasificación internacional de los síndromes epilépticos diferencia la epilepsia, desde un punto de vista etiopatogénico, en idiopática (de causa desconocida, con factores genéticos involucrados y buen pronóstico inicial), criptogénica (de causa no establecida, presumiblemente orgánica y con un pronóstico incierto) y sintomática (secundarias a lesiones y con un pronóstico frecuentemente desfavorable). Las principales causas de crisis epilépticas se exponen en la tabla 12,5.

El pediatra deberá conocer cuáles son las principales causas de crisis epilépticas y mostrar una especial
atención ante fenómenos paroxísticos observados en pacientes con antecedentes familiares de epilepsia, pacientes con retraso mental, niños con trastornos neurocutáneos...
Factores genéticos
La predisposición genética desempeña un papel muy importante en las convulsiones febriles y las epilepsias idiopáticas. En algunos casos se ha determinado la localización de los genes implicados en la epilepsia; así, el brazo largo del cromosoma 20 y el brazo corto del cromosoma 6 alojan los genes involucrados en las convulsiones neonatales familiares benignas y la epilepsia mioclónica juvenil, respectivamente. En otros síndromes epilépticos, los antecedentes familiares frecuentes son los que evidencian la predisposición genética; así, encontramos antecedentes familiares positivos de epilepsia en el 35% de los pacientes con epilepsia mioclónico-atónica, en el 15-45% de los niños con ausencias infantiles, en un 25% de casos si se trata de ausencias
mioclónicas, etc.
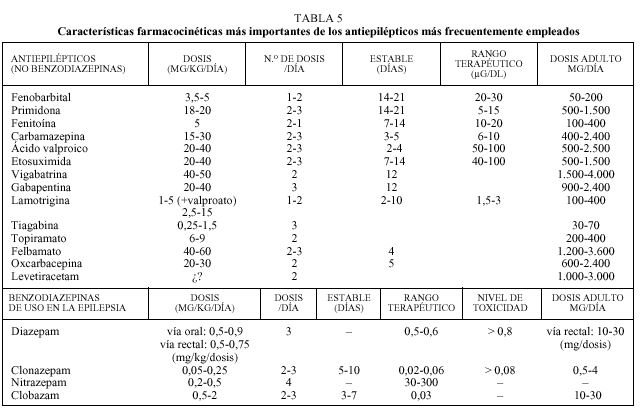
Otro grupo de enfermedades genéticamente determinadas asocian las crisis epilépticas como una manifestación clínica característica del trastorno. Este grupo incluye los trastornos neurocutáneos (p. ej., epilepsia en el 90, el 50 y el 80% de los pacientes con esclerosis tuberosa, hipomelanosis de Ito o síndrome de Sturge-Weber, respectivamente)6 (fig. 1), las epilepsias mioclónicas progresivas (enfermedad de Lafora, sialidosis...), las enfermedades mitocondriales y un amplio grupo de enfermedades de origen metabólico (aminoacidopatías, glucogenosis...).
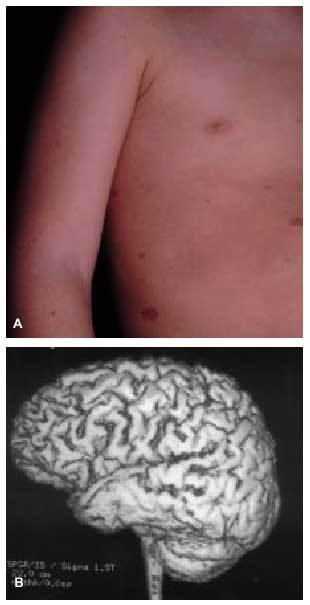
Fig. 1. A) Manchas café con leche en varón con dificultades de aprendizaje y epilepsia. B) RM cerebral de reconstrucción tridimensional, que revela la presencia de polimicrogiria del lóbulo temporal del mismo paciente.
Un grupo menor de pacientes con factores de riesgo de clara involucración genética serían aquellos trastornos que asocian la epilepsia con una mayor prevalencia respecto a la población general. Así, el 5-10% de los pacientes con síndrome de Down son epilépticos, según algunas series, el 20-40% de los niños con síndrome X frágil, etc.
Factores adquiridos
Dentro del grupo de factores prenatales debemos resaltar las malformaciones cerebrales (paquigiria, esquisencefalia...) (fig. 2), los accidentes cerebrovasculares intrauterinos, las infecciones por microorganismos neurotropos (citomegalovirus, toxoplasmosis...) o las encefalopatías tóxicas.
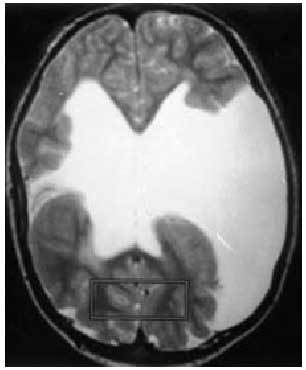
Fig. 2. Esquisencefalia bilateral en paciente con déficit motor y epilepsia.
Entre los factores perinatales, la encefalopatía hipóxico-isquémica representa la causa más frecuente de crisis neonatales. Otros trastornos incluyen las hemorragias intra y periventriculares, más frecuentes en el prematuro, la sepsis neonatal, diferentes metabolopatías o alteraciones metabólicas...
La causa de la epilepsia, en un último grupo de pacientes, se basará en el antecedente de meningoencefalitis, traumatismos craneoencefálicos graves, tumores cerebrales o malformaciones vasculares... La presencia de infecciones que afectan al SNC es una de las causas más frecuentes de epilepsia en la edad pediátrica, a diferencia de lo que se observa en edades posteriores. En relación con los traumatismos craneoencefálicos (TCE), la gravedad del mismo se asocia con una frecuencia diferente de epilepsia o crisis tardías; en los TCE leves, el riesgo de crisis en los primeros 5 años de vida no supera el 0,6%, frente al 11% en las formas graves. Del mismo modo, la presencia de hematomas intracraneales, convulsiones precoces o fractura craneal con hundimiento inciden desfavorablemente en estas frecuencias. Por último, el hallazgo de una malformación arteriovenosa o un tumor cerebral como causa de crisis epilépticas en un niño es, afortunadamente, poco frecuente. Sin embargo, debemos señalar que más de la mitad de los casos con cualquiera de estas dos enferme-
dades presentarán crisis convulsivas, en ocasiones como
primera manifestación clínica del proceso.
Diagnóstico clínico de la primera crisis epiléptica
Un 10-15% de los niños va a presentar a lo largo de su vida un trastorno paroxístico, el 20% serán convulsiones febriles, el 10% crisis epilépticas y el resto fenómenos paroxísticos no epilépticos. La valoración clínica inicial, efectuada con frecuencia por el pediatra de atención primaria, deberá identificar la tipología clínica, realizar un correcto diagnóstico diferencial y dirigir una primera aproximación diagnóstica7-9.

En la historia clínica debemos recoger las características del episodio, tras solicitar a los padres que simulen la crisis: posibles circunstancias asociadas, pródromos, relación con el ritmo vigilia-sueño, movimientos acompañantes (revulsión ocular, automatismos...), duración del episodio y características de la poscrisis.
Con estos datos podremos apuntar inicialmente la tipología clínica de la crisis (tabla 2).

Hay que conocer los fenómenos paroxísticos no epilépticos para descartarlos10. La mayor parte de estos trastornos los podremos descartar simplemente con la
historia clínica. El síncope vasovagal es el error diagnóstico más frecuente, especialmente en la infancia; éste se relaciona a menudo con el ortostatismo, el
paciente presenta palidez y sudación, el inicio suele
ser gradual, la pérdida de conciencia no supera el minuto, rara vez se observan movimientos anormales asociados o relajación de esfínteres y la recuperación es generalmente completa en pocos minutos. En niños menores otro fenómeno a descartar es el espasmo del sollozo, pálido o cianótico, que se precede siempre por factores muy característicos (un traumatismo leve, un episodio frustrante...). Otros trastornos paroxísticos a descartar son los síncopes cardíacos, los terrores nocturnos, los tics, las mioclonías benignas del sueño, el reflujo gastroesofágico (síndrome de Sandifer), las seudocrisis...
Es obligado anotar de forma completa los antecedentes personales (control del embarazo, características del parto, infecciones del SNC, convulsiones febriles...). Se debe acompañar de un cuidadoso interrogatorio sobre el desarrollo psicomotor (la epilepsia es 2-3 veces más frecuente en niños con retraso) y de enfermedades no neurológicas (cardiopatía, nefropatía...).
Los antecedentes familiares son importantes, tanto de epilepsia como de retraso mental, sordera, etc.
La historia clínica se completará con una detallada exploración física. Debe recoger el aspecto físico, el desarrollo ponderostatural, los rasgos dismórficos (incluso aquellos que nos parezcan sutiles), el comportamiento durante la entrevista, las discromías y las visceromegalias. En la exploración neurológica no podemos olvidar medir el perímetro cefálico y estudiar el fondo de ojo.
Una vez llegada a este punto, si el pediatra sospecha que el paciente ha sufrido una crisis epiléptica, debe remitirle a la consulta de neuropediatría; esta derivación se realizará igualmente en pacientes bien controlados que presenten una crisis aislada. En general, los estudios complementarios abordables desde la atención primaria aportarán poca información adicional. Los estudios neurofisiológicos y radiológicos deberán ser recomendados y valorados por el neurólogo infantil; la presencia de alteraciones paroxísticas focales o generalizadas en el EEG no implica siempre el diagnóstico de epilepsia. En otros casos, el trazado es enormemente indicativo de la enfermedad (síndrome de West, ausencias...) (fig. 3).
Fig. 3. EEG: trazado característico de la epilepsia de ausencias con punta onda a 3 Hz.
En general, más del 50% de los niños presentan una sola crisis convulsiva si no tienen factores de riesgo de recurrencia (tabla 3). La presencia de algunos de estos factores, como la edad menor de dos años, la exploración neurológica anormal, la presencia de crisis de larga duración, la sospecha clínica de un factor etiológico de mal pronóstico o una gran angustia familiar justifican la derivación preferente o urgente a un hospital para su ingreso y estudio. Una mayor frecuencia de crisis en pacientes epilépticos bien controlados justifica igualmente la derivación urgente del paciente.

Características farmacocinéticas y farmacodinámicas
En la última década se han introducido diversos fármacos antiepilépticos (FAE) que contribuyen notablemente al control clínico del paciente epiléptico. En contrapartida, obligan a los profesionales médicos infantiles a conocer las indicaciones del fármaco, sus características, los posibles efectos colaterales y las interacciones del mismo.
Indicaciones
Indudablemente, la decisión de administrar un FAE concreto, el momento de realizarlo y el tiempo más adecuado para su retirada es una función que no le corresponde, en principio, al pediatra. Sin embargo, es necesario que conozca las indicaciones básicas de cada uno de ellos, ya que actualmente están siendo debatidos los protocolos terapéuticos de primera y segunda elección en la epilepsia (tabla 4).

Características
El pediatra de atención primaria debe conocer algunas de las características farmacocinéticas más importantes de cada antiepiléptico (tabla 5)11-13.
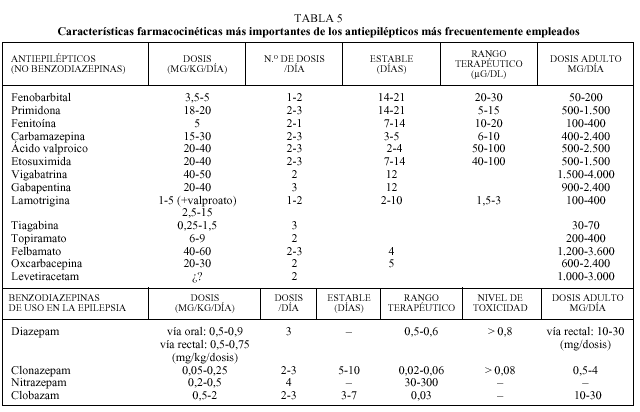
Al igual que con la mayor parte de las medicaciones empleadas en pediatría, la dosis debe ajustarse inicialmente al peso del paciente. La dosis inicial suele ser una cuarta o quinta parte de la dosis de mantenimiento, debiéndose aumentar la dosificación cada 3-7 días hasta alcanzar la dosis final.
Para algunos fármacos disponemos de un rango terapéutico, definido como aquella concentración plasmática de un sustancia antiepiléptica con la cual el 80% de los pacientes está libre de crisis y no manifiesta efectos tóxicos; este parámetro tan sólo es orientativo, ya que varía según el laboratorio, la gravedad del síndrome epiléptico o ante el empleo de varios fármacos de forma simultánea.
La eliminación de los fármacos se realizará por vía renal, hepática o mixta. Esta eliminación justifica el
ajuste de la medicación en situaciones como la insuficiencia hepática o renal. Algunos fármacos, como fenitoína, carbamazepina, valproato, etosuximida, lamotrigina, tiagabina y benzodizepinas, se eliminan por vía hepática; la vigabatrina y la gabapentina se eliminan por por vía renal; el fenobarbital, la primidona, el topiramato y el felbamato utilizan ambas vías para su eliminación.
Efectos secundarios
Los efectos tóxicos de los FAE engloban aquellos fenómenos neurotóxicos relacionados con el propio mecanismo del fármaco (sedación, ataxia...), efectos teratogénicos, a los que no haremos mención por las características del paciente, y los efectos idiosincráticos, dependientes en gran medida de la fisiología del niño que recibe el tratamiento (hipersensibilidad, hepatotoxicidad...).
La mayor parte de los efectos neurotóxicos aparecen al inicio del tratamiento, a menudo desaparecen con el tiempo, o se potencian o inician con la introducción de otro fármaco. Estos efectos obligan a retirar la medicación excepcionalmente (5-10%).

Los fenómenos idiosincráticos engloban las alteraciones del tejido conectivo (carbamazepina, fenobarbital...), las alteraciones dermatológicas (carbamazepina, lamotrigina...), las discrasias sanguíneas (carbamazepina, felfamato, fenitoína...), la hepatotoxicidad (carbamazepina, valproato...). Estos efectos idiosincráticos son potencialmente letales y obligan a retirar la medicación.
Resumimos a continuación los efectos secundarios más frecuentes de los antiepilépticos más empleados y las recomendaciones para algunos de ellos (tabla 6).

Fenobarbital (y primidona)
Los efectos secundarios más importantes se relacionan con su neurotoxicidad, e incluyen sedación, nistagmo, ataxia, disartria, alteraciones del comportamiento, depresión, irritabilidad e hipercinesia. Produce depleción de vitamina K, vitamina D y ácido fólico.
Los efectos idiosincráticos engloban las reacciones exantemáticas, incluido el síndrome de Stevens-Johnson, la hepatotoxicidad y las alteraciones del tejido conectivo tipo reumático, como la fibrosis plantar, la artritis y la contractura de Dupuytren.
Es recomendable valorar el aporte adicional de vitaminas y ácido fólico durante primer año de vida.
Fenitoína
La neurotoxicidad de la fenitoína es claramente inferior a la del fenobarbital cuando se encuentra en rango terapéutico. Los efectos estéticos como la hiperplasia gingival, el hirsutismo o las alteraciones del tejido conectivo, limitan el empleo crónico de esta medicación en la infancia. Con valores superiores a 20-30 µg/dl pueden aparecer diplopía, ataxia, nistagmo, disartria, asociando incluso una atrofia cerebelosa irreversible en tratamientos mantenidos.
Las reacciones idiosincráticas engloban los exantemas, las discrasias sanguíneas, el síndrome lupus-like y la hepatotoxicidad. Pueden también interferir en el metabolismo de las vitaminas referidas en el apartado anterior.
Entre las recomendaciones, se debe señalar la necesidad de una higiene bucal extrema y los controles estrechos de la dosificación empleada por su absorción errática y su cinética no lineal.
Carbamazepina
Los efectos sobre el SNC de esta medicación están relacionados con su concentración en sangre. Puede producir somnolencia, vértigo, astenia, diplopía o temblor. Son también habituales la dispepsia y las náuseas, aunque con frecuencia son transitorias y tolerables. Otros fenómenos colaterales menos frecuentes son la neutropenia leve asintomática y la hiponatremia dilucional secundaria al efecto agonista renal de la ADH (poco frecuente en niños).
Las reacciones idiosincráticas engloban de nuevo las reacciones exantemáticas (fig. 4), incluyendo el síndrome de Stevens-Johnson y el síndrome de Lyell, la aplasia medular, el lupus eritematoso y la hepatotoxicidad, aunque estos dos últimos son muy infrecuentes.
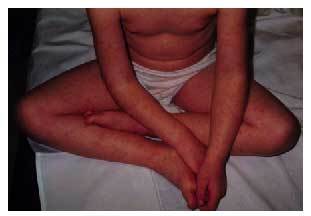
Fig. 4. Exantema maculopapuloso en paciente tratado con carbamazepina.
El empleo de la carbamazepina se debe controlar de forma analítica (hemograma y función hepática). La leucopenia intensa (< 2.000 celulas/µl) y mantenida (2%), o la elevación de las transaminasas por encima de 150 U/l obliga, generalmente, a la retirada de la medicación.
Ácido valproico
La neurotoxicidad de este antiepiléptico es escasa, describiéndose casos con temblor, sedación o inquie tud motriz. Los efectos colaterales gastrointestinales, como las náuseas y los vómitos, son los efectos secundarios dependientes de la dosis más frecuentes, y de forma habitual, escasamente limitantes. Otros efectos adversos incluyen la caída del cabello, la ganancia ponderal en el tratamiento crónico o las irregularidades menstruales, habiéndose señalado una mayor frecuencia de poliquistosis ovárica. Esta última circunstancia ha sido referida recientemente, y precisa una mayor consistencia científica según nuestro criterio y experiencia.
Las reacciones idiosincráticas incluyen una hepatotoxicidad grave, más frecuente en menores de 2 años con politerapia. Otras reacciones poco frecuentes son la plaquetopenia, la pancreatitis hemorrágica o la depleción de carnitina.
La presencia de dolor abdominal en un niño en tratamiento con ácido valproico obliga la determinación de la función hepática y la amilasemia. La trombopenia por debajo de 100.000 cél/µl o la hipertransaminemia persistente obligan a retirar el tratamiento.
Benzodiazepinas
La neurotoxicidad de este grupo de fármacos suele ser transitoria y consiste, predominantemente, en sedación, mareos o ataxia. En niños puede asociarse a hiperexcitabilidad paradójica o dificultades del aprendizaje. Del mismo modo, puede favorecer la retención o el aumento de las secreciones bronquiales. La administración intravenosa puede inducir depresión cardiorrespiratoria.
Su empleo obliga a controlar los procesos respiratorios, debiéndose valorar el uso de mucolíticos.
Interacciones farmacológicas
Las interacciones de los FAE son frecuentes, lo que obliga a menudo al control de los valores terapéuticos. La evaluación de las interacciones entre diferentes antiepilépticos corresponde al neurólogo infantil; sin embargo, el pediatra debe conocer cómo pueden interactuar los antiepilépticos mayores con fármacos de uso habitual en estos niños.
El fenobarbital y la fenitoína actúan como inductores enzimáticos acelerando el metabolismo de otros antiepilépticos, anticoagulantes, antibióticos, cimetidina, teo-
filina, ciclosporina, corticoides, anticonceptivos y algunas vitaminas liposolubles. El ácido fólico disminuye los valores de ambos FAE.
Los macrólidos, la isoniazida, los antagonistas del calcio y la fluoxetina aumentan los valores de carbamazepina. Este fármaco favorece la inducción enzimática, como la fenitoína y el fenobarbital, por lo que puede disminuir también la concentración y la eficacia de algunos de los fármacos descritos previamente.
Las sulfamidas, la isoniazida, el alopurinol, el omeprazol, la cimetidina, el cloranfenicol y la fluoxetina aumentan los valores plasmáticos de la fenitoína. La rifampicina reduce los valores de la misma. El alcohol, ingerido de forma aguda, puede aumentar también sus valores.
Los salicilatos reducen la concentración total de fenitoína y el ácido valproico, aumentando su concentración libre.
El alcohol, los antidepresivos, los antihistamínicos o los descongestivos nasales potencian los efectos neurotóxicos del fenobarbital y las benzodiazepinas.
Controles periódicos clínicos y analíticos
El paciente epiléptico precisa controles periódicos, incluso con independencia del tratamiento empleado, para vigilar la evolución clínica de sus crisis convulsivas, los fenómenos colaterales, como las dificultades de aprendizaje o la adaptación del mismo o su familia a la epilepsia14.
Controles clínicos
El primer control clínico debe realizarse al mes o mes y medio, si se ha instaurado tratamiento farmacológico. Posteriormente, recomendamos controles cada 3-12 meses dependiendo del paciente, su edad o el tipo de epilepsia.
Con la introducción de la medicación debemos vigilar la tolerancia y sus posibles efectos secundarios. En estos controles nos aseguraremos del rendimiento académico y la asistencia escolar. En niños de menor edad estableceremos un control de su desarrollo psicomotor. De forma periódica, anotaremos el desarrollo ponderostatural.
Una vez alcanzada la dosis de mantenimiento, se valorará la eficacia del mismo, anotando la desaparición de las crisis o, por el contrario, su persistencia y frecuencia.
En tratamientos prolongados, se vigilará la calidad de vida del paciente epiléptico, la situación ambiental del mismo, así como las relaciones sociales y familiares. Para este propósito, podemos hacer uso de diferentes escalas de calidad de vida, algunas especialmente diseñadas para la población epiléptica, como el cuestionario Cave15. Dentro del manejo multidisciplinario de la epilepsia, el psiquiatra o el psicólogo infantil pueden favorecer los mecanismos adecuados de ayuda, educación y consejo.
Controles analíticos
Existe una gran controversia en la necesidad de los controles periódicos analíticos, especialmente en relación con las determinaciones de los valores plasmáticos. Esta disyuntiva se plantea por la escasa correlación entre los valores plasmáticos y los efectos terapéuticos o tóxicos de algunos FAE16. La monitorización sistemática de fenitoína, fenobarbital, primidona, carbamazepina y etosuximida es generalmente útil, o al menos conveniente. La determinación periódica de las cifras de ácido valproico, lamotrigina, gabapentina o benzodiazepinas revela escasa información en relación con posibles efectos terapéuticos o tóxicos.
Por lo general, la mayoría de los autores recomiendan la determinación del valor plasmático del fármaco al inicio del tratamiento, 1-4 semanas después de alcanzar la dosis de mantenimiento, ante la aparición de efectos tóxicos o sospecha de incumplimiento terapéutico, al añadir otro antiepiléptico, con la aparición de enfermedades intercurrentes (hepáticas o renales), en situaciones asociadas al cambio del metabolismo del fármaco (ganancia ponderal, adolescencia...) o de forma previa a la retirada del tratamiento.
La extracción de la muestra sanguínea para la determinación los valores plasmáticos se recomendará antes de la primera dosis de la mañana.
Igualmente, se realizará un hemograma y una función hepática básica, una o dos veces por año, siendo preciso el control más estricto en el manejo de algunos antiepilépticos, como el felbamato, o en situaciones concretas, como el empleo del ácido valproico en menores de 2 años.
Recomendaciones generales para la vida diaria del paciente epiléptico
El propósito principal del abordaje multidisciplinario de estos pacientes debe ser la preservación de un modo de vida similar al de cualquier otro niño. A continuación, anotaremos unas recomendaciones generales que el pediatra debe conocer17.
Desde el punto de vista escolar, los niños epilépticos obtienen generalmente peores calificaciones, según refleja la mayor parte de las series; esta circunstancia puede estar motivada por la propia epilepsia, el tratamiento, el origen de la epilepsia o los factores psicosociales asociados (temor del profesor, sobreprotección de los padres...)18. Sin embargo, esto no es una norma constante; por ello el paciente con crisis bien controladas o de escasa frecuencia, y un nivel de inteligencia normal, debe llevar una escolarización normal. Si durante el proceso aparecen dificultades de aprendizaje, éstas deben ser diagnosticadas y tratadas precozmente. Los profesores o cuidadores deben conocer la enfermedad del niño y saber cómo actuar ante una crisis.
Cuando las crisis aparecen o persisten durante la adolescencia, éstas pueden motivar diferentes trabas laborales. Indudablemente, existen profesiones desaconsejadas para estas pacientes, como la conducción o el pilotaje de aviones. Según la recomendación de la ILAE, para obtener el carné de conducir, es preciso la ausencia de crisis al menos durante 2 años.
La actividad física es siempre aconsejable. Favorece la salud general del paciente y mejora las relaciones sociales. En relación con los deportes, algunos deben ser supervisados de forma próxima, como la natación. En el ciclismo deben extremarse las medidas de seguridad (casco, acompañantes...); otros, como el submarinismo, el motociclismo o el alpinismo, son desaconsejables.
La televisión y los videojuegos son habitualmente tolerables. Sin embargo, en la epilepsia con crisis fotosensibles deben limitarse estos entretenimientos; se tomarán precauciones como mantener la habitación iluminada, ver la televisión a una distancia de 2,5-3 m, evitar mirarla cuando se apaga o enciende y hacer uso de protectores de pantalla.
Es obligado que el paciente duerma entre 7 y 10 h, evitando los despertares precoces innecesarios, e intentando mantener la mayor regularidad posible en los hábitos de sueño.
No se debe modificar la dieta en estos niños, ya que no se ha constatado un mejor control de las crisis con ninguna de estas medidas. La dieta cetógena podría ser útil en el tratamiento de niños con epilepsias refractarias. En la actualidad, su empleo es escaso por su mal cumplimiento y tolerancia, así como la existencia de un arsenal terapéutico cada vez mayor19.
El tabaco (aunque desaconsejado en la edad pediátrica) no incrementa la frecuencia de convulsiones. Sin embargo, el consumo de alcohol está claramente contraindicado en el paciente epiléptico por su potencial epileptógeno y sus interacciones con los FAE20.
Finalmente, debemos recordar que la menstruación o los embarazos pueden aumentar la frecuencia de las crisis en el paciente epiléptico.
Actuación ante situaciones especiales
El riesgo de convulsión por vacunación o fiebre es discretamente mayor. Parece prudente evitar el componente celular antipertussis de la vacuna triple DTP. No existen contraindicaciones para otras vacunas.
Si el paciente vomita en la primera media hora después de la toma, deberá administrarse de nuevo. Si los vómitos persisten, debe fraccionarse la misma en varias tomas para favorecer la tolerancia oral.
Las enfermedades intercurrentes, la propia afección o los tratamientos empleados pueden interferir desfavorablemente en el control de las crisis. Debe realizarse un control estricto en enfermedades que afecten directamente la función hepática o renal, por las propias características farmacodinámicas de los FAE.
La mayor parte de las intervenciones quirúrgicas no repercuten notablemente en la administración periódica de la medicación antiepiléptica. En los casos en los que se precise una dieta absoluta prolongada se optará por administrar la medicación por vía intravenosa o rectal cuando esta circunstancia sea posible, o por la administración rectal de benzodiazepinas de forma profiláctica hasta que pueda instaurarse de nuevo la medicación habitual por vía oral.